化學誘導后白木香轉錄組文庫的構建與測序
2013-09-14 12:47:48吳宏清王磊陶美華高曉霞白玲章衛民
生物技術通報 2013年8期
關鍵詞:研究
吳宏清 王磊 陶美華 高曉霞 白玲 章衛民
白木香[Aquilaria sinensis(Lour.)Gilg]又稱土沉香,瑞香科沉香屬植物,是國產沉香的唯一植物資源[1]。當白木香樹干受到物理、化學傷害或真菌侵染的情況下,可分泌出一種氣味芬芳的防御性黑色樹脂,即為我國傳統名貴藥材沉香。在自然界,沉香的形成需要幾年至十幾年、甚至數十年的時間。為了快速獲得珍貴的沉香藥材,人們通過各種方法人工造香。布蘭切特和范貝克[2]利用亞硫酸氫鈉、氯化鈉、甲酸等化學物質誘導沉香屬(Aquilaria spp.)植物產生沉香;Chen等[3]用氯化鈉誘導成年白木香產生的沉香與天然沉香的化學成分極為相似。倍半萜類化合物是沉香的主要藥效成分[4],Kumeta和 Ito[5]及 Okudera 和 Ito[6]的研究結果表明,水楊酸和茉莉酸甲酯能誘導沉香屬植物的懸浮細胞產生沉香倍半萜前體物質α-愈創木烯(α-guaiene)、α-蛇麻烯(α-humulene)和 δ-愈創木烯(δ-guaiene)。Xu等[7]從傷害誘導的白木香細胞中成功克隆到主產物為δ-愈創木烯的合成酶基因,但是從前體物質到沉香特征產物間的代謝途徑仍然未知。
轉錄組測序是最近發展起來的利用深度測序進行轉錄組分析的技術[8],目前已廣泛應用于生物學、醫學和臨床研究及藥物研發等。轉錄組測序結合表達譜分析的方法,可以在沒有參考基因組的條件下對轉錄組進行全測序,以獲得的轉錄組信息為參考,對不同樣品的表達譜進行基因注釋,通過比較獲得不同表達譜間的差異表達基因,進而研究其基因功能。因此,利用轉錄組測序有助于白木香結香相關功能基因的發現,闡明沉香特征產物的代謝途徑,揭示人工誘導白木香結香的分子機制。
本研究對5年樹齡、化學誘導后1年的成年白木香植株進行總RNA的提取,獲得的總RNA用于Illumina轉錄組測序,經測序文庫的構建,上機測序,數據過濾,序列組裝,旨在獲得完整的白木香轉錄組信息,為后續的表達譜分析積累基礎數據。
1 材料與方法
1.1 材料
1.1.1 試驗材料 試驗樣品采自廣東省信宜市珍稀沉香發展有限公司的白木香基地,參考王磊等[9]進行結香試驗,其中用于轉錄組測序的白木香樣品取自同一株5年樹齡、甲酸處理后1年的已結香的白木香植株及另外一株未進行結香試驗的白木香植株。樣品采集時利用前端成鉤狀的刨刀,除去樹皮后,在樹干上從外到內依次刨下樣品,外圍未變色部分為白木樣品(W樣品),與白木相鄰的一圈深棕色木材為結香樣品(A樣品),白木樣品與結香樣品間還有部分顏色為淺棕色的木質部,作為結香與未結香間的過渡樣品(T樣品),最內側已腐爛的部分作為腐木樣品(D樣品),以及從另外一株未結香植株上采集的白木樣品(C樣品)。采集完樣品后立即用錫箔紙包裹置于液氮中保存。
1.1.2 主要儀器與試劑 BioSpec-nano生命科學紫外/可見分光光度計;EPS 601電泳儀;GE ImageQuant 350凝膠成像系統;Hettich VNIVERSAL-32R臺式冷凍離心機;Agilent 2100生物分析儀;Illumina HiSeqTM2000測序儀。焦碳酸二乙酯(DEPC)購自廣州杰順生物科技有限公司;改良異硫氰酸胍-CTAB提取液(38%水飽和酚,1mol/L異硫氰酸胍,2% CTAB,100mmol/L NaAc-HAc pH5.2,2mol/L NaCl,2% PVP),用前混勻;抽提液Ⅰ(水飽和酚∶氯仿∶異戊醇 = 25∶24∶1);抽提液Ⅱ(氯仿∶異戊醇 = 24∶1)。
1.2 方法
1.2.1 白木香總RNA的提取 采用改良異硫氰酸胍-CTAB法分別提取白木香W、A、T、D和C樣品總RNA:樣品用液氮研磨后迅速分裝到含有改良異硫氰酸胍-CTAB提取液的離心管中,劇烈震蕩,室溫靜置5min;等體積的抽提液I抽提2次,離心取上清;等體積的抽提液II抽提1次,離心取上清;加入1/2體積的無水乙醇以及與上清等體積的4mol/L LiCl,顛倒混勻,-30℃靜置過夜,離心取沉淀;沉淀溶于適量DEPC處理水中,加入1/10體積3mol/L NaAc-HAc,混勻后,加入3倍體積無水乙醇,-30℃靜置30min,4℃離心10min;75%乙醇洗滌沉淀2次,溶于30μL DEPC處理水中低溫保存。Agilent 2100生物分析儀對總RNA的RIN值及28S∶18S比值進行檢測,驗證RNA的完整性。等量合并各樣品總RNA進入下一步操作。
1.2.2 白木香轉錄組測序文庫的構建 用帶有Oligo(dT)的磁珠富集總RNA樣品中的mRNA,加入fragmentation buffer將mRNA打斷成短片段,以打斷后的mRNA為模板,用六堿基隨機引物合成第一條cDNA鏈。加入緩沖液、dNTPs、RNase H和DNA polymerase I合成第二條cDNA鏈,然后用QiaQuick PCR純化試劑盒純化產物,用EB緩沖液洗脫后做末端修復、加A并連接測序接頭,然后用瓊脂糖凝膠電泳進行片段大小選擇,最后進行PCR擴增,建好的測序文庫用Illumina HiSeqTM2000進行測序。
1.2.3 Illumina HiSeqTM2000上機測序[11]使用 Illumina HiSeqTM2000測序平臺進行轉錄組文庫的測序。樣品為白木香W、A、T、D和C各樣品合并后的轉錄組測序文庫。測序得到的原始圖像經base calling轉化為序列數據Raw reads。
1.2.4 數據過濾 對測序所得的Raw reads進行過濾,濾去的數據包括含接頭的reads,N的比例大于5%的reads,重復的和質量數較低的reads(質量值Q≤10的堿基數占整個read的20%以上),過濾后所得為Clean reads,后續分析都基于此Clean reads。
1.2.5 序列的 De novo 組裝[12]使用 Trinity[10]軟件對Clean reads做De novo組裝。將具有一定長度overlap的reads連成更長的片段Contig,然后與Clean reads重新比對,通過paired-end reads確定Contig所屬的轉錄本以及在轉錄本中的分布,Trinity軟件能將這些Contig連在一起,得到兩端不能再延長的序列。然后使用Tgicl對其進行去冗余和進一步拼接,并對其進行同源轉錄本聚類,得到最終的Unigene。聚類后Unigene分為兩部分,一部分是clusters(以CL開頭),另一部分是singletons(以Unigene開頭)。
2 結果
2.1 白木香總RNA的獲得
使用改良異硫氰酸胍-CTAB法提取白木香各組織總RNA,經Agilent 2100生物分析儀檢測,RIN值最小為6.7,28S∶18S均大于1.0,RNA總量遠大于20μg,滿足轉錄組測序的需求,結果如表1所示。

表1 用于轉錄組測序的白木香總RNA質量
2.2 測序產量統計
如表2所示,Illumina HiSeqTM2000上機測序后獲得Raw reads共58804828條,過濾后獲得Clean reads共54685634條,總測序長度為4921707060nt,Q20值達97.45%,測序質量較高。
2.3 組裝結果統計及質量評估
如表3所示,經初步組裝后,共獲得190109條Contigs,平均長度有324nt,N50值為549,進一步組裝后,共獲得83467條Unigenes,平均長度高達702nt,N50值較高,達1120,序列組裝理想,使得白木香的轉錄組數據得到較好的保存。

表2 白木香轉錄組測序統計

表3 組裝結果
圖1為所獲得Contigs的組裝統計結果。序列長度大于等于500nt的Contigs高達26786條,占總Contigs的14.09%;其中,大于等于1000nt的Contigs達2518條,占總Contigs的1.32%;大于等于2000nt的Contigs達1429條,占總Contigs的0.75%;大于等于3000nt的Contigs有1012條,占總Contigs的0.53%。
圖2表示Contigs進一步組裝后獲得Unigenes的組裝統計結果。序列長度大于等于1000nt的Unigenes高達17155條,占總Unigenes的20.56%;其中,大于等于2000nt的Unigenes有5189條,占總Unigenes的6.22%;大于等于3000nt的Unigenes有1691條,占總Unigenes的2.03%。
3 討論
轉錄組測序技術是建立在新一代高通量測序平臺(如Roche GS FLX或Illumina HiseqTM2000)上的cDNA測序技術,自2008年Nature和Science上分別發表利用轉錄組測序技術研究裂殖酵母(Schizosaccharomyces pombe)、釀酒酵母(Saccharomyces cerevisiae)轉錄組的論文[13,14]后,轉錄組測序技術已成為研究轉錄組的革命性工具。對比轉錄組學研究的其他方法,轉錄組測序可檢測任意物種的轉錄組,無需前提信息,無需克隆,理論上可檢測所有轉錄信息。

圖1 白木香轉錄組Contigs組裝統計
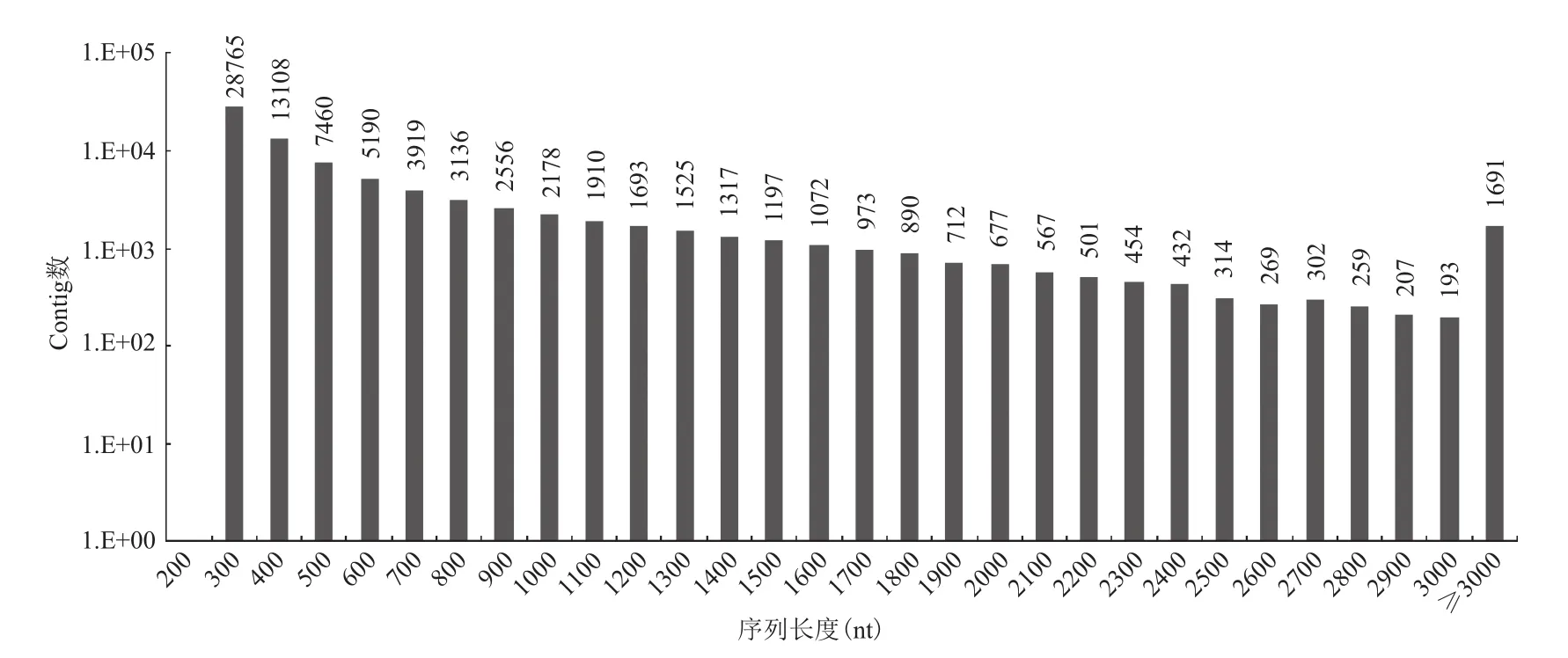
圖2 白木香轉錄組Unigenes組裝統計
為了研究白木香未結香組織和結香組織間的表達差異,獲得與結香相關的功能基因,可以采用數字基因表達譜分析的方法,篩選未結香與結香組織間的差異表達基因。然而,如今NCBI上未有白木香的參考基因組或參考轉錄組信息,因此必須先進行轉錄組測序,獲得總的轉錄組信息,在獲得轉錄組序列的基礎上進行表達譜分析及差異基因的研究。
張爭等[15]利用454測序平臺,對機械傷害后的白木香莖的轉錄組進行測序,共獲得22095條平均長度為314nt的Unigenes。本研究對化學誘導后白木香樣品的轉錄組測序、組裝,共獲得83467條平均長度為702nt的Unigenes,轉錄組信息保存較完整,為化學誘導白木香結香機理的研究提供大量的基礎數據。為了便于后續的表達譜分析,防止因個體差異引起的無關信息過多,本研究選擇來自同一株白木香不同部位的W、T、A和D 4個樣品用于后續的分析,可消除因來自不同植株的樣品對差異基因篩選的干擾。將W樣品作為對照組,T、A和D 3個樣品分別作為試驗組,獲取各對照的差異表達基因,研究其基因功能,進而揭示化學誘導白木香結香的分子機理,獲得與化學誘導相關的基因和轉錄因子。
4 結論
采用改良異硫氰酸胍-CTAB法提取白木香各組織總RNA,構建轉錄組測序文庫后進行Illumina HiSeqTM2000上機測序,共獲得54685634條Clean reads,總長度為4921707060nt,經多次組裝獲得83467條Unigenes,平均長度為702nt,N50值為1120,大于等于3000nt的Unigenes有1691條,占總Unigenes的2.03%,測序和組裝質量較高。
[1]中國科學院中國植物志編輯委員會. 中國植物志[M]. 北京:科學出版社, 1999:290.
[2]布蘭切特RA, 范貝克HH. 栽培的沉香木:US, ZL 02810500.1[P]. 2004-10-27.
[3]Chen HQ, Yang Y, Xue J, et al. Comparison of compositions and antimicrobial activities of essential oils from chemically stimulated agarwood, wild agarwood and healthyAquilaria sinensis(Lour.)Gilg trees[J]. Molecules, 2011, 16(6):4884-4896.
[4]楊俊山. 沉香化學成分的研究概況[J]. 天然產物研究與開發,1998, 10(1):99-103.
[5]Kumeta Y, Ito M. Characterization of δ-guaiene synthases from cultured cells ofAquilaria,responsible for the formation of the sesquiterpenes in agarwood[J]. Plant Physiol, 2010, 154(4):1998-2007.
[6]Okudera Y, Ito M. Production of agarwood fragrant constituents inAquilaria calliand cell suspension[J]. Plant Biotechnology, 2009,26(3):307-315.
[7]Xu YH, Zhang Z, Wang MX, et al. Identification of genes related to agarwood formation:transcriptome analysis of healthy and wounded tissues ofAquilaria sinensis[J]. BMC Genomics, 2013, 14(1):227.
[8]祁云霞, 劉永斌, 榮威恒. 轉錄組研究新技術:RNA-Seq及其應用[J]. 遺傳 , 2011, 33(11):1191-1202.
[9]王磊, 章衛民, 高曉霞, 等. 一種人工誘導白木香產生沉香的方法 :中國,CN102302041A[P]. 2012-01-04.
[10]Grabherr MG, Haas BJ, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome[J].Nat Biotechnol, 2011, 29(7):644-652.
[11]Feng C, Chen M, Xu CJ, et al. Transcriptomic analysis of Chinese bayberry(Myrica rubra)fruit development and ripening using RNA-Seq[J]. BMC Genomics, 2012, 13:19-33.
[12]Xiang LX, He D, Dong WR, et al. Deep sequencing-based transcriptome profiling analysis of bacteria-challengedLateolabrax japonicusreveals insight into the immune relevant genes in marine fish[J]. BMC Genomics, 2010, 11:472-492.
[13]Wilhelm BT, Marguerat S, Watt S, et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution[J]. Nature, 2008, 453(7199):1239-1243.
[14]NagalakshmiU, Wang Z, Waern K, et al. The transcriptional landscape of the yeast genome defined by RNA sequencing[J].Science, 2008, 320(5881):1344-1349.
[15]張爭, 高志暉, 魏建和, 等. 三年生白木香機械傷害轉錄組學研究[J]. 藥學學報 , 2012, 47(8):1106-1110.
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
遼金歷史與考古(2019年0期)2020-01-06 07:45:20
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年11期)2018-08-04 03:26:04
汽車工程學報(2017年2期)2017-07-05 08:13:02
國際商務財會(2017年8期)2017-06-21 06:14:14
電子制作(2017年23期)2017-02-02 07:17:19
