吡啶基離子液體與噻吩類化合物相互作用的理論研究
2013-10-22 06:23:16呂仁慶王淑濤
石油學報(石油加工) 2013年6期
關鍵詞:結構
呂仁慶,林 進,王淑濤
(1.中國石油大學 理學院化學系,山東 青島266580;2.中國石油大學 化學工程學院,山東 青島266580)
在室溫或室溫附近呈液態的由離子構成的物質稱為室溫離子液體。2001年,Bosmann等[1]首先發現,離子液體可有效地脫除汽油中的含硫化合物。2003年,Lo等[2]采用H2O2氧化和離子液體萃取的聯合方法脫除汽柴油中的含硫化合物,發現比單一的離子液體萃取脫硫更為有效。Anantharaj等[3-9]采用模擬方法考察了咪唑基離子液體與含硫化合物的相互作用。近年來,吡啶基離子液體的脫硫研究受到關注[10-20]。吡啶基離子液體脫除噻吩類化合物的效果較好,普遍推測由π…π相互作用所致。2007年,Wang等[10]考察了N-丁基吡啶醋酸鹽離子液體([BPY][Ac])的脫硫作用。筆者擬采用密度泛函理論優化篩選了噻吩(TS)、苯并噻吩(BT)、二苯并噻吩(DBT)、萘(NAP)與N-丁基吡啶醋酸鹽離子液體([BPY][Ac])相互作用的結構,采用NBO和AIM分析,在分子水平上證實了TS、BT、DBT、NAP與[BPY][Ac]的氫鍵作用、π…π作用等本性。
1 吡啶基離子液體與芳香化合物相互作用的計算方法
研究氫鍵體系時,采用密度泛函理論(DFT)比Hartree-Fock方法更可靠,并且所用優化時間遠遠小于MP2方法。采用 MS中DMol3軟件包[21-22]的GGA/PW91泛函[23]和DNP基組對所有初始結構進行了非限制性全優化。盡管PW91泛函不能很好地描述色散作用,但GGA/PW91/DNP可以給出很好的優化結果[24]。最穩定結構的頻率分析表明沒有虛頻。對最穩定的結構進行了 NBO[25]和 AIM[26-27]分析。
由于DMol3采用的是數字函數(Numerical functions),比傳統的Gaussian函數計算所得的基組重疊誤差(BSSE)要小,所以可以忽略[28]。將[BPY][Ac]、TS/BT/DBT/NAP的能量與[BPY][Ac]-TS/BT/DBT/NAP能量差定義為相互作用能,其計算如式(1)所示。
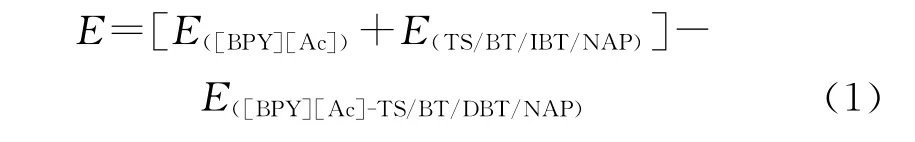
2 結果與討論
2.1 [BPY][Ac]-TS/BT/DBT/NAP的優化結構及電子性質
為了尋找到最穩定的[BPY][Ac]結構,首先優化了獨立的[BPY]+陽離子和[Ac]-陰離子結構,并對其進行靜電勢分析。結果表明,[BPY]+陽離子的正電性主要集中在靠近N的吡啶環上和丁基上的C—H基團,而[Ac]-陰離子的負電性主要集中在2個O原子上。在設計[BPY][Ac]初始結構時,考慮了[BPY]+陽離子的正電性較高區域和[Ac]-陰離子的負電性較高區域發生相互作用,由于[BPY]+陽離子不具有對稱性,考慮了[Ac]-陰離子在 N與丁基兩側的相互作用的情況。GGA/PW91/DNP全優化[BPY][Ac]最穩定結構如圖1(a)所示。[Ac]-陰離子在 H2、H71一側能量最低、最穩定,而在H6、H72一側的能量相對較高。[Ac]-陰離子中的O1與吡啶環上的C2—H2發生相互作用,其作用距離為0.1721nm;其它相互作用,即H71…O2和H91…O2的作用距離分別為0.1873和0.2889nm,而H和O的范德華半徑(0.120nm 和0.152nm)之和為0.272nm[29]。
[BPY][Ac]-TS的最穩定的結構如圖1(b)所示。由圖1(b)可知,[Ac]-陰離子與[BPY]+陽離子相互作用距離,即H2…O1、H71…O1、H81…O2的作用距離分別為0.1958、0.2329、0.2483nm,均小于它們的范德華半徑之和[29]。而噻吩處于吡啶環的上方,TS環和吡啶環彼此平行,TS與[BPY][Ac]相互作用距離,即H1’…O2、S…C4、C2’…C5的作用距離分別為0.2166、0.3548、0.3630nm。TS與[BPY][Ac]的相互作用不僅有H…O氫鍵,且有S…C4、C2’…C5作用,意味著有π…π相互作用。所謂π…π相互作用,又稱為π…π堆積(π…πstacking),即芳香環的非共價鍵(Non-covalent)相互作用[30-31]。盡管π…π相互作用經常發生,但是還沒有一個統一的觀點來解釋這種現象,因為影響π…π相互作用的因素復雜,如靜電作用(四極-四極、四極-偶極、偶極-偶極作用)、疏水作用、van der Waals相互作用,且在不同的情況下不同因素所起的作用不同。π…π相互作用主要有4種形式,分別為夾心型、T型、平行位錯型和斜交型[30-34]。TS環和吡啶環平行,但發生一定的位錯,屬于平行位錯型。一般來說,氣相狀態下,很難形成芳香環平行的π…π相互作用,但是H1’…O2的相互作用有助于芳香環的平行相互作用[35]。
圖1(c)為優化的最穩定[BPY][Ac]-BT 結構。從圖1(c)可知,[Ac]-陰離子位于吡啶環的C2—H2附近,并與丁基發生作用,陰、陽離子之間的相互作用距離,即H2…O1、H81…O1、H71…O2、H91…O2的作用距離分別為0.1785、0.2886、0.1889、0.2791nm。BT與[BPY][Ac]的相互作用距離,即H4’…O1、H3’…O1的作用距離分別為0.2680、0.2280nm,均明顯小于它們的范德華半徑之和。BT環和吡啶環的相互作用為斜交型,C3’…H3距離較短,為0.2768nm,存在著π…π相互作用。
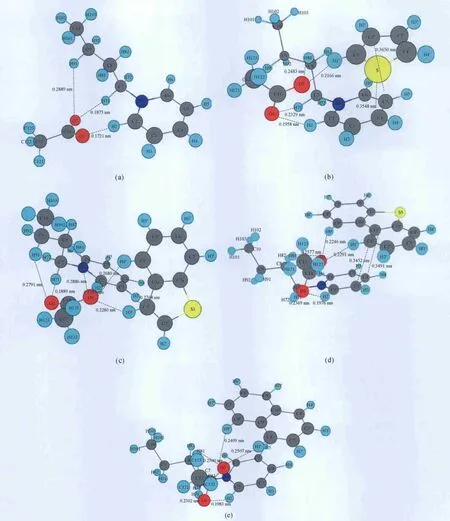
圖1 [BPY][Ac]、[BPY][Ac]-TS、[BPY][Ac]-BT、[BPY][Ac]-DBT和[BPY][Ac]-NAP優化結構和相互作用距離示意圖Fig.1 Scheme of optimized structures and some interacting distances for[BPY][Ac],[BPY][Ac]-TS,[BPY][Ac]-BT,[BPY][Ac]-DBT and[BPY][Ac]-NAP
[BPY][Ac]-DBT 的最穩定結構如圖1(d)所示。從圖1(d)可知,[Ac]-陰離子位于[BPY]+陽離子的C2—H2和丁基之間。[BPY][Ac]與DBT相互作用距離,即H9’…O2、H1’…O2、C1’…C3和C11’…C4的作用距離分別為0.2246、0.2291、0.3452和0.3491nm。DBT環與吡啶環相互平行,并且有一定的錯位,可以推測發生了π…π相互作用。
為了考察[BPY][Ac]離子對與其它芳香化合物的相互作用,優化篩選了[BPY][Ac]-NAP的最穩定結構,如圖1(e)所示。從圖1(e)可知,[BPY][Ac]與NAP相互作用距離,即H8’…O2、H1’…O2的作用距離分別為 0.2409、0.2507nm。比較[BPY][Ac]與DBT、NAP的相互作用可知,DBT環和NAP環均平行于吡啶環,作用方式類似,均為O2與吸附分子中的2個H發生作用,但DBT形成的氫鍵作用距離短,這可能是由于DBT環上的H1’和H9’的正電性比NAP上的 H1’和H8’的正電性大所致。DBT的H1’和H9’的正電性分別為+0.23873 和 +0.23873,[BPY][Ac]-DBT 的H1’和 H9’的正電性分別為+0.28202 和+0.28148;NAP的 H1’和 H8’的正電性分別為+0.23632 和 +0.23632,[BPY][Ac]-NAP 的H1’和 H8’的正電性分別為+0.27461 和+0.27219。NAP環和吡啶環的距離較大,沒有明顯的C…C相互作用。
在上述5個最穩定的結構中,由H2形成的氫鍵的距離最短、作用最大,可能是由于吡啶環上的N原子的電負性相對較大,吸電子性較強,導致C2—H2上的正電荷較高,故形成的氫鍵最強,與ZHU等[36]的研究結果一致。上述結構性質表明,TS、BT、DBT與[BPY][Ac]發生了π…π相互作用。
2.2 [BPY][Ac]-TS/BT/DBT/NAP的相互作用能ΔE
按照式(1)計算得到[BPY][Ac]和 TS、BT、DBT、NAP相互作用能分別為44.14、57.55、65.58和30.51kJ/mol,相互作用能從大到小的順序為[BPY][Ac]和DBT、[BPY][Ac]和 BT、[BPY][Ac]和TS、[BPY][Ac]和 NAP。同為芳香化合物,但TS及其衍生物與離子對的相互作用明顯比NAP大,可能是由于其較強的極性和較強的π…π相互作用所致。
2.3 [BPY][Ac]-TS/BT/DBT/NAP穩定結構的NBO分析
表1為[BPY][Ac]、[BPY][Ac]-TS、[BPY][Ac]-BT、[BPY][Ac]-DBT 和[BPY][Ac]-NAP中的授-受相互作用及其二級微擾穩定化能(E(2))。E(2)反映電子授-受相互作用的強度,E(2)越大,電子授-受相互作用程度大,相互作用強。由表1可知,在[BPY][Ac]、[BPY][Ac]-TS、[BPY][Ac]-BT、[BPY][Ac]-DBT、[BPY][Ac]-NAP 中,LP(O1σ*(C2—H2)的E(2)分別為142.12、45.44、116.33、38.99 和 44.39kJ/mol。由此可見,H2所形成的氫鍵作用較強。對比表2相互作用距離可知,LP(O)(C—H)之間的E(2)越大,其相互作用的距離越短。
由NBO分析可知,在吡啶環上N原子周圍上的正電性高于其它原子,這是由于N的電負性較大,吸引電子的能力較強,導致其周圍正電性明顯高于其它原子上的正電性,因而O原子與這些H原子形成的氫鍵強,這與靜電勢分析結果一致。
由表1還可知,[BPY][Ac]-TS的最穩定結構中不僅有氫鍵(O…H—C)作用,還有π…π、π…H—C、π…O—C相互作用,并且S原子明顯參與了TS與[BPY][Ac]的相互作用(LP(S)σ(C4—C3));[BPY][Ac]-BT的最穩定結構中有氫鍵(O…H—C)、π…π、π…H—C 相互作用;[BPY][Ac]-DBT的最穩定結構中有氫鍵(O…H—C)、π…π、O…π、π…H—C相互作用;[BPY][Ac]-NAP的最穩定結構中有氫鍵(O…H—C)、π…π、O…π、π…H—C相互作用。由此可見,不僅TS、BT、DBT與吡啶環有π…π相互作用,NAP環和吡啶環也有π…π相互作用,但由于噻吩類化合物的芳香環上的富電子性和極性,與吡啶環相互作用較強。TS、BT、DBT上S原子的NBO電荷分別為+0.45527、+0.43055、+0.42110,盡管 TS中的S的正電性最高,但只有其與[BPY][Ac]有較強的相互作用(LP(Sσ(C4—C3)),這可能是BT和DBT的空間位阻較大造成的。
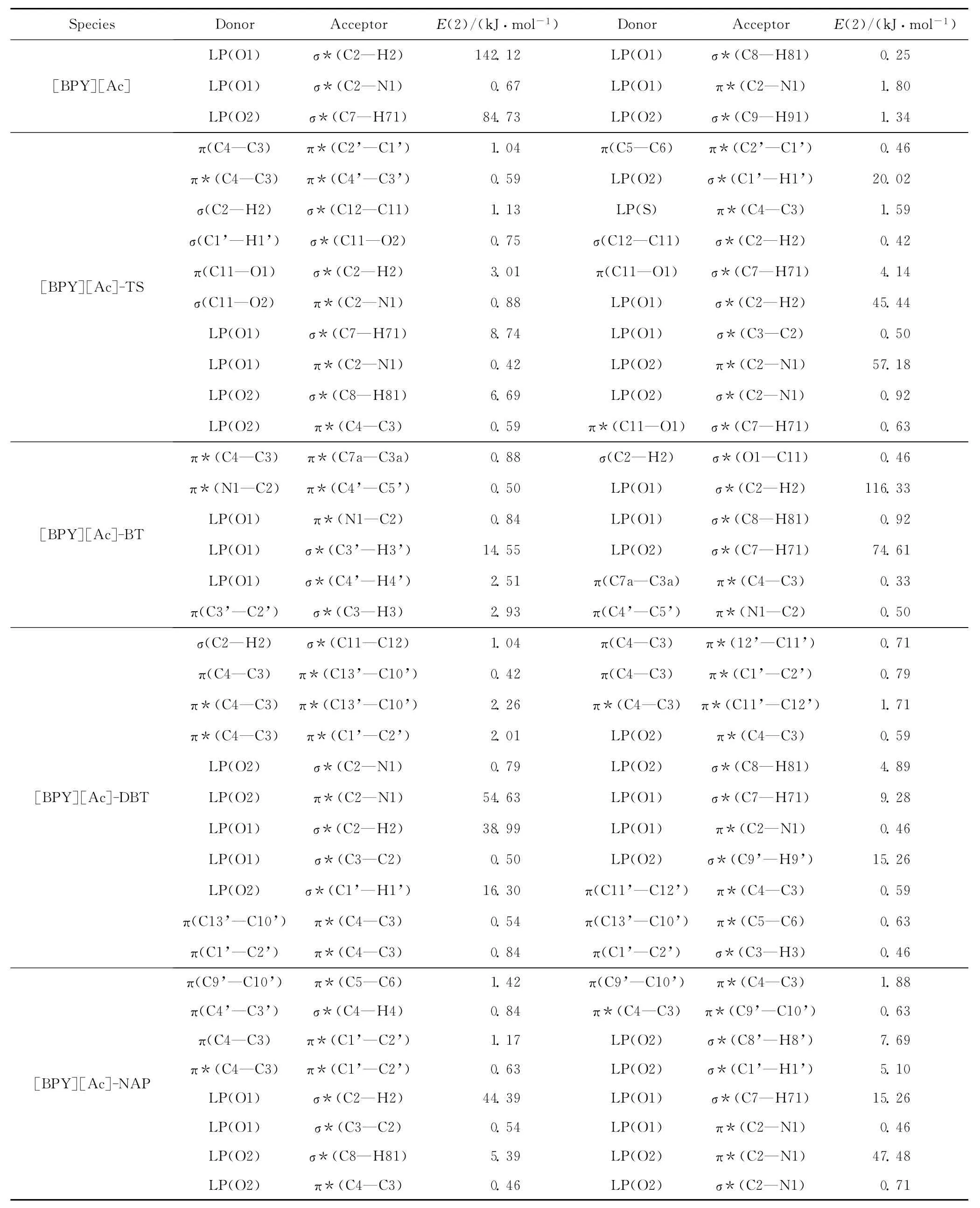
表1 [BPY][Ac]、[BPY][Ac]-TS、[BPY][Ac]-BT、[BPY][Ac]-DBT、[BPY][Ac]-NAP中的授-受相互作用及其二級微擾穩定化能(E(2))Table 1 Some donor-acceptor interactions in[BPY][Ac],[BPY][Ac]-TS,[BPY][Ac]-BT,[BPY][Ac]-DBT,[BPY][Ac]-NAP and their second order perturbation stabilization energies(E(2))
2.4 [BPY][Ac]-TS/BT/DBT/NAP穩定結構的拓撲性質分析
電子密度的拓撲學分析主要是采用Bader所提出的分子內原子(Atoms in Molecules,AIM)的理論[37]。所有臨界點的▽2ρ均為正值,說明這些弱相互作用均為閉殼相互作用。表2為[BPY][Ac]、[BPY][Ac]-TS、[BPY][Ac]-BT、[BPY][Ac]-DBT、[BPY][Ac]-NAP的電子密度(ρ)和拉普拉斯量(▽2ρ)。 由表2可見, [BPY][Ac]-TS 中[BPY][Ac]與TS相互作用分別為O2…H2’、C2’…C5;[BPY][Ac]-BT中[BPY][Ac]與BT相互作用分別為 O1…H3’、O1…H4’、C3’…H3;[BPY][Ac]-DBT中[BPY][Ac]與 DBT相互作用分別為O2…H1’、O2… H9’、C1’…C3、C11’…C4;[BPY][Ac]-NAP 中 [BPY][Ac]和NAP相互作用分別為O2…H1’、O2…H8’。從AIM分析可知,TS、BT、DBT與吡啶環有明顯的π…π相互作用,與[Ac]-陰離子有氫鍵作用,與NBO分析結果相一致。而NAP與[BPY][Ac]中的陰離子有氫鍵作用,NAP環和吡啶環之間沒有出現C…C相互作用的臨界點,說明π…π相互作用較弱。
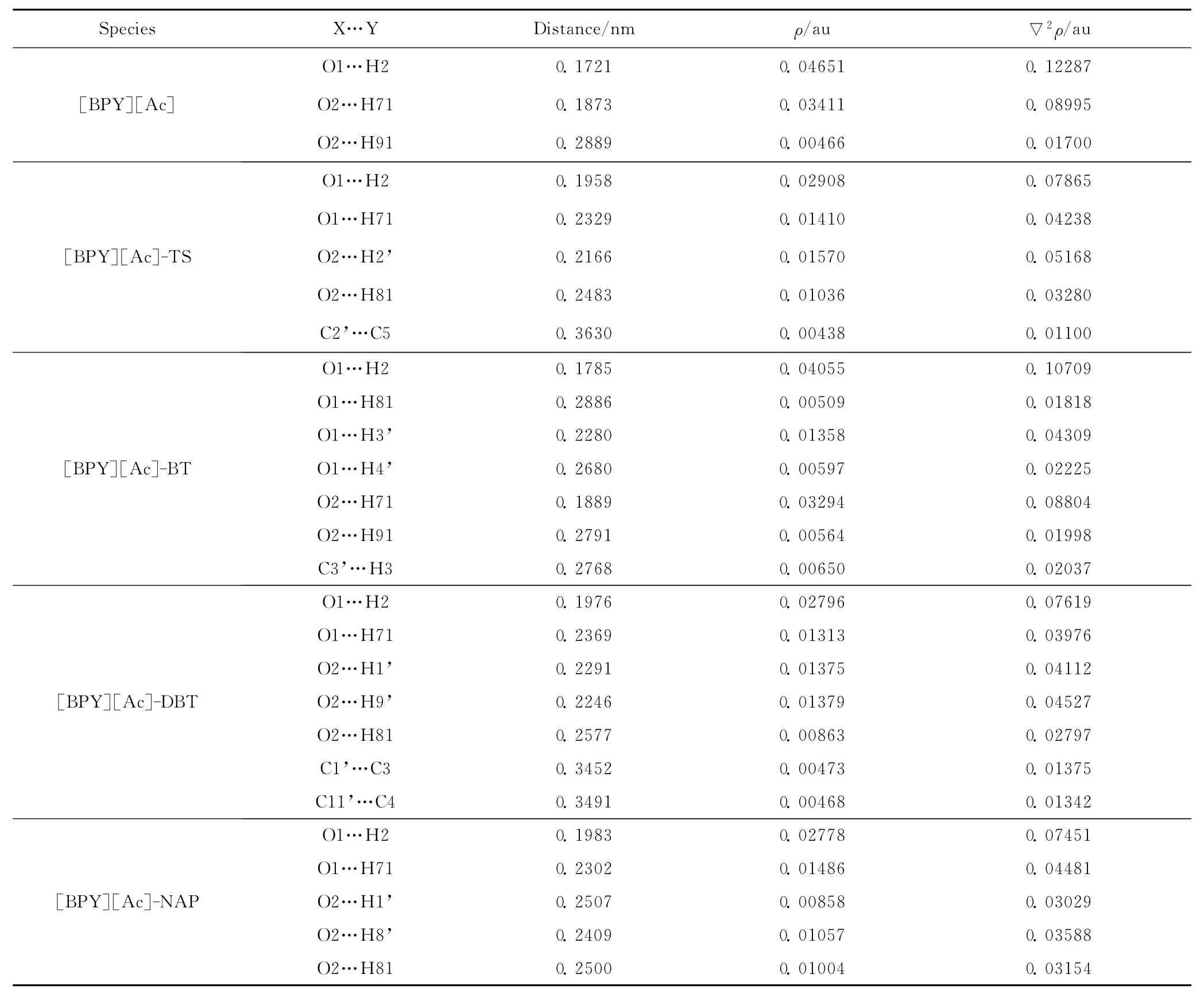
表2 [BPY][Ac]、[BPY][Ac]-TS、[BPY][Ac]-BT、[BPY][Ac]-DBT、[BPY][Ac]-NAP的電子密度(ρ)和拉普拉斯量(▽2ρ)Table 2 The topological properties of electron density(ρ)and Laplacian of density(▽2ρ)of[BPY][Ac],[BPY][Ac]-TS,[BPY][Ac]-BT,[BPY][Ac]-DBT,and[BPY][Ac]-NAP
3 結 論
采用GGA/PW91/DNP方法非限制性的全優化了TS、BT、DBT、NAP、[BPY][Ac]、[BPY][Ac]-TS、[BPY][Ac]-BT、[BPY][Ac]-DBT、[BPY][Ac]-NAP的結構,對最穩定的結構進行了NBO和 AIM 分析。[BPY][Ac]、[BPY][Ac]-TS、[BPY][Ac]-BT、[BPY][Ac]-DBT、[BPY][Ac]-NAP的最穩定結構顯示,[Ac]-陰離子總是優先靠近吡啶基中的C2-H2,這是由于吡啶環上N的電負性較大,造成C2-H2的正電性較高所致。在分子水平上,證實了[BPY][Ac]與噻吩、苯并噻吩、二苯并噻吩、萘的相互作用中有氫鍵、π…C—H、π…π相互作用。[BPY][Ac]與噻吩、苯并噻吩、二苯并噻吩、萘的相互作用能分別為44.14、57.55、65.58和30.51kJ/mol,其中,萘與[BPY][Ac]相互作用較弱。
[1]BOSMANN A,DATSEVICH L,JESS A,et al.Deep desulfurization of diesel fuel by extraction with ionic liquids [J]. Chemical Communications, 2001,2494-2495.
[2]LO W,YANG H,WEI G.One-pot desulfurization of light oils by chemical oxidation and solvent extraction with room temperature ionic liquids [J]. Green Chemistry,2003,5:639-642.
[3]ANANTHARAJ R,BANERJEE T.Phase behavior of 1-ethyl-3-methylimidazolium thiocyanate ionic liquid with catalytic deactivated compounds and water at several temperatures:Experiments and theoretical predictions[J].International Journal of Chemical Engineering,2011,2011(1):1-13.
[4]KUMAR A A P,BANERJEE T.Thiophene separation with ionic liquids for desulphurization:A quantum chemical approach [J].Fluid Phase Equilibria,2009,278(1-2):1-8.
[5]ANANTHARAJ R, BANERJEE T. Liquid-liquid equilibria for quaternary systems of imidazolium based ionic liquid+thiophene+pyridine+iso-octane at 298.15K: Experiments and quantum chemical predictions[J].Fluid Phase Equilibria,2011,312:20-30.
[6]SANTIAGO R S,SANTOS G R, AZNAR M.UNIQUAC correlation of liquid-liquid equilibrium in systems involving ionic liquids:The DFT-PCM approach[J].Fluid Phase Equilibria,2009,278(1-2):54-61.
[7]HANKE C G,JOHANSSON A,HARPER J B,et al.Why are aromatic compounds more soluble than aliphatic compounds in dimethylimidazolium ionic liquids?A simulation study[J].Chemical Physics Letters,2003,374(1-2):85-90.
[8]KEDRA-KROLIK K,FABRICE M,JAUBERT J.Extraction of thiophene or pyridine fromn-heptane using ionic liquids,gasoline and diesel desulfurization [J].Industrial &Engineering Chemistry Research,2011,50(4):2296-2306.
[9]ANANTHARAJ R,BANERJEE T.Quantum chemical studies on the simultaneous interaction of thiophene and pyridine with ionic liquids[J].AIChE Journal,2011,57(3):749-764.
[10]WANG J,ZHAO D,ZHOU E,et al.Desulfurization of gasoline by extraction withN-alkyl-pyridinium based ionic liquids [J].Journal of Fuel Chemistry and Technology,2007,35(3):293-296.
[11]GAO H,LUO M,XING J,et al.Desulfurization of fuel by extraction with pyridinium-based ionic liquids[J].Industrial & Engineering Chemistry Research,2008,47(21):8384-8388.
[12]ZHAO D, WANG Y, DUAN E. Oxidative desulfurization of fuel oil by pyridinium based ionic liquids[J].Molecules,2009,14(11):4351-4357.
[13]GAO H,LI Y,WU Y,et al.Extractive desulfurization of fuel using 3-methylpyridinium-based ionic liquids[J].Energy Fuels,2009,23(5):2690-2694.
[14]FRANCISCO M,ARCE A,SOTO A.Ionic liquids on desulfurization of fuel oils[J].Fluid Phase Equilibria,2010,294(1-2):39-48.
[15]ZHAO D,WANG Y,DUAN E,et al.Oxidation desulfurization of fuel using pyridinium based ionic liquids as phase transfer catalysts[J].Fuel Processing Technology,2010,91(12):1803-1806.
[16]VERDíA P,GONZáLEZ E J,RODRíGUEZ-CABO B,et al.Synthesis and characterization of new polysubstituted pyridinium-based ionic liquids:Application as solvents on desulfurization of fuel oils[J].Green Chemistry,2011,13:2768-2776
[17]ANANTHARAJ R, BANERJEE T. Fast solvent screening for the simultaneous hydrodesulfurization and hydrodenitrification of diesel oil using ionic liquids[J].Journal of Chemical Engineering Data,2011,56 (6):2770-2785.
[18]GAO H,GUO C,XING J,et al.Deep desulfurization of diesel oil with extraction using pyridinium-based ionic liquids[J].Separation Science Technology,2012,47(2):325-330
[19]ZHANG C,PAN X,WANG F,et al.Extraction oxidation desulfurization by pyridinium-based task specific ionic liquids[J].Fuel,2012,102:580-584.
[20]RODRIGUEZ-CABO B,FRANCISCO M,SOTO A,et al.Hexyl dimethylpyridinium ionic liquids for desulfurization of fuels.Effect of the position of the alkyl side chains [J].Fluid Phase Equilibria,2012,314:107-112.
[21]DELLEY B. An all-electron numerical method for solving the local density functional for polyatomic molecules[J].Journal of Chemical Physics,1990,92(1):508-517.
[22]DELLEY B.From molecules to solids with the DMol3approach[J].Journal of Chemical Physics,2000,113(18):7756-7764.
[23]PERDEW J P,WANG Y.Accurate and simple analytic representation of the electron-gas correlation energy[J].Physical Review B,1992,45(23):13244-13249.
[24]CASTELLANO O, GIMON R, SOSCUN H.Theoretical study of theσ—πandπ—πinteractions in heteroaromatic monocyclic molecular complexes of benzene,pyridine,and thiophene dimers:Implications on the resin-asphaltene stability in crude oil[J].Energy Fuels,2011,25(6):2526-2541.
[25]REED A E, CURTISS L A, WEINHOLD F.Intermolecular interactions from a natural bond orbital,donor-acceptor viewpoint[J].Chemical Reviews,1988,88(6):899-926.
[26]BIEGLER-K?NIG F,SCH?NBOHM J.Update of the AIM2000program for atoms in molecules[J].Journal of Computational Chemistry,2002,23(15):1489-1494.
[27]BIEGLER-K?NIG F,SCH?NBOHM J,BAYLES D.AIM2000——A program to analyze and visualize atoms in molecules[J].Journal of Computational Chemistry,2001,22(5):545-559.
[28]INADA Y,ORITA H.Efficiency of numerical basis sets for predicting the binding energies of hydrogen bonded complexes:Evidence of small basis set superposition error compared to Gaussian basis sets[J].Journal of Computational Chemistry,2008,29 (2):225-232.
[29]BONDI A.Van der Waals volumes and radii [J].Journal of Physical Chemistry,1964,68(3):441-451.
[30]SINNOKROT M O,VALEEV E F,SHERRILL C D.Estimates of the ab initio limit forπ—πinteractions:The benzene dimmer[J].Journal of American Chemical Society,2002,124(36):10887-10893.
[31]HUNTER C A,SANDERS J K M.The nature ofπ—π interactions[J].Journal of American Chemical Society,1990,112(14):5525-5534.
[32]COCKROFT S L,HUNTER C A,LAWSON K R,et al.Electrostatic control of aromatic stacking interactions[J].Journal of American Chemical Society,2005,127(24):8594-8595.
[33]SINNOKROT M O,SHERRILL C D.Unexpected substituent effects in face-to-faceπ-stacking interactions[J].Journal of Physical Chemistry A,2003,107(41):8377-8379.
[34]RINGER A L,SINNOKROT M O,LIVELY R P,et al.The effect of multiple subsitituents on sandwich and T-shaped pi-pi interactions [J].Chemistry-A European Journal,2006,12(14):3821-3828.
[35]RASHKIN M J, WATERS M L. Unexpected substituent effects in offsetπ—πstacked interactions in water[J].Journal of American Chemical Society,2002,124(9):1860-1861.
[36]ZHU X,CUI P,ZHANG D,et al.Theoretical study for pyridinium-based ionic liquid 1-ethylpyridinium trifluoroacetate: Synthesis mechanism, electronic structure,and catalytic reactivity [J].Journal of Physical Chemistry A,2011,115(29):8255-8263.
[37]BADER R F W. A quantum theory of molecular structure and its applications [J].Chemical Reviews,1991,91(5):893-928.
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
