基于糖苷芳香酸酯的金屬有機材料的制備與研究
2014-07-14 05:19:36楊瑞環霍冀川
無機化學學報 2014年5期
楊瑞環 張 馳*,,2 霍冀川 曲 鵬
(1西南科技大學材料科學與工程學院,綿陽 621010)
(2新西蘭奧克蘭大學化學科學學院,奧克蘭 1142,新西蘭)
0 引 言
金屬有機骨架化合物(metal organic frameworks,簡稱MOFs)是一種新型的多孔材料,因其具有高孔性、比表面積大、合成方便、骨架規模大小可變以及可根據目標要求作化學修飾、結構豐富等優點[1],在氣體吸附分離[2-3]、催化劑[4-6]、磁性材料[7]、光學材料[8]和藥物釋放[9]等領域受到人們的廣泛關注,已經成為材料化學領域中的一個研究熱點[10-12]。
MOFs是由含氧、氮等的多齒有機配體(大多是芳香多酸和多堿)與金屬離子或金屬簇通過自組裝的方式,由金屬離子或金屬簇作為頂點,經過剛性的或半剛性的有機配體連接而成的網狀骨架結構[8,13]。1995年由Yaghi教授[14]首次合成了MOF-5儲氫材料,并在2003年Science雜志上報道了測試結果。2005年,Yaghi教授[15]通過改變MOF-5的有機聯結體制備了一系列IRMOFs材料,分析和研究它們的儲氫性能。Millward和Yaghi等[16]測得MOF-177對CO2的吸附能力明顯高于沸13X和活性炭粉末等傳統材料。Snejko等[17]綜合了磺酸的強酸性和稀土元素的催化活性這兩個因素,利用1,5-二磺酸萘的鈉鹽(NDS)與 Ln(NO3)36H2O(ln=La、Pr和 Nd)通過水熱合成得到3種配位化合物,即MOFs催化材料。劉麗麗等[18]采用合成后共價修飾法和一鍋法分別制備了催化劑IRMOF-3-SI-Au(PS)和IRMOF-3-SI-Au(OP)探索了這兩種催化劑在醛、炔和胺三組分偶聯反應中的催化性能。Qiu研究小組報道的JUC-48材料[19],在它的六邊形的孔道中組裝了激光染料分子:羅丹明6G,組裝后的熒光性能較為理想,且可隨溫度變化,充分說明MOFs材料在熒光領域大有可為。近年來,一些稀土元素也用于合成MOFs材料[20]。由此可見,MOFs材料已經被廣大科研工作者所重視。
甲基葡萄糖苷是一種用途廣泛的有機原料,它是一種帶有獨特環狀結構的四羥基多元醇,具有非離子型表面活性劑的屬性[21]。該糖苷分子上含有四個羥基官能團,很容易與羧酸等發生反應來達到改性的目的。只要改性的基團本身具有孤對電子或含有游離電子的官能團,該化合物就可能與過渡金屬離子配位。通過分子重排和自組裝最終形成一種具有拓撲狀結構的金屬配合物。目前,關于將生物質材料改性后的新型有機配體用于金屬有機材料制備的報道還很少[22-23]。
本文以自然界普遍存在的生物質材料——甲基葡萄糖苷作為基體,借助化學改性,將含有孤對電子的齒狀特征官能團引入到糖苷分子中,合成芳香酸酯,并采用MOFs材料的制備方法和配位原理,將芳香酸酯與金屬鹽溶液配位來制備具有一定比表面積的材料。主要以制得的MGAE作為有機配體,通過擴散法在模板劑三乙胺的作用下與不同的金屬離子配位來制備一系列金屬配合物,并對制得的配合物進行較全面的表征。
1 實驗部分
1.1 實驗材料
甲基葡萄糖苷,阿拉丁化學有限公司;硝酸鎘,硝酸鉛,醋酸鋅,三乙胺,鄰苯二甲酸酐,丙酮,分析純,成都科龍化工試劑廠;氯化鋅,分析純,成都市聯合化工試劑研究所;蒸餾水,實驗室自制。
1.2 儀器設備
101型電熱鼓風干燥箱,北京市永光明醫療儀器廠;Cp225D電子計數天平,德國Sartorius公司;DF-101S集熱式恒溫加熱磁力攪拌器,江蘇金壇儀器廠;Spectrum One型傅立葉變換紅外光譜儀(KBr壓片,測試范圍:500~4 000 cm-1),美國;SDT Q600同步熱分析儀(N2氣氛,流量:100 mL·min-1,升溫速率 10℃·min-1,溫度范圍:室溫~ 650 ℃),美國 TA公司;X′Pert PRO X 射線衍射儀(掃描角度 3°~80°,銅靶,電壓:40 KV;電流:40 mA),荷蘭帕納科公司;Ultra 55場發射掃描電鏡(表面鍍金,電壓:15 KV),德國蔡司公司;Oxford IE450X-Max80能譜儀,牛津儀器公司;NOVA 3000比表面積測試儀(100℃條件下熱處理2 h,77 K條件下氮氣吸附脫附),美國康塔公司。
1.3 MGAE-金屬配合物的制備
1.3.1 MGAE 的合成
稱取鄰苯二甲酸酐23 g將其倒入三口燒瓶中,加入100 mL丙酮,在50℃條件下攪拌使其完全溶解,之后加入10 g甲基葡萄糖苷,將溫度升至70℃后加入10 mL三乙胺作為催化劑,混合溶液在攪拌條件下回流反應8 h,白色渾濁溶液逐漸變為透明的無色液體,冷卻至室溫后,使用無水硫酸鎂干燥,過濾,所得濾液在60℃條件下攪拌加熱除去丙酮溶劑,得到粘稠的無色液體產物。所得產物經硅膠板(石油醚和丙酮作為展開劑,V石油醚/V丙酮=1∶1)點板,證明產物中不含鄰苯二甲酸酐。
1.3.2 配合物的制備
稱取25 g的MGAE溶解在120 mL的丙酮中。分別稱取15 g硝酸鎘,硝酸鉛,氯化鋅和醋酸鋅溶解在100 mL蒸餾水中。分別量取20 mL MGAE溶液和20 mL金屬鹽溶液將其混合,攪拌條件下逐滴加入三乙胺來調節溶液的酸堿度,隨著三乙胺的加入,混合溶液逐漸有固體析出,將混合溶液pH值調至7~8后在室溫條件下攪拌4 h使其充分配合。所得混合溶液經過濾后得到的固體產物依次用蒸餾水和丙酮洗滌數次,之后置于60℃烘箱中干燥至恒重。
2 結果與討論
2.1 FT-IR和EDS的結果分析
MGAE及其與硝酸鎘、硝酸鉛、氯化鋅和醋酸鋅等金屬鹽溶液形成的配合物的紅外光譜圖如圖1所示。MGAE在3 500 cm-1和3 400 cm-1之間有一寬而強的峰,此峰是羥基的特征吸收峰,可能是甲基葡萄糖苷分子上未完全反應的羥基或者游離的羧基上的羥基吸收峰;3 000 cm-1左右處的吸收峰為甲基葡萄糖苷分子上的甲基吸收峰;1 727 cm-1處的吸收峰是羰基的特征吸收峰;1 601、1 584、1 487和1 452 cm-1對應于苯環的特征吸收峰;1 272 cm-1是碳氧鍵的吸收峰;而在1 000 cm-1到1 200 cm-1之間的雙肩峰是糖環的特征吸收峰[24]。與MGAE的紅外光譜圖相比,配合物在1 727 cm-1附近的吸收峰幾乎被完全抑制,表明羰基在配位過程中參與了反應,同時,圖2的配合物能譜圖中所含的金屬元素說明羰基與金屬元素發生了反應。圖1b中(Pb-MGAE配合物)羰基的特征吸收峰未被完全抑制,可能是由于鉛離子與MGAE未完全配位。所得的配合物在1 400 cm-1到1 600 cm-1之間的吸收峰是苯環的特征吸收峰,說明所得配合產物中含有苯環結構。
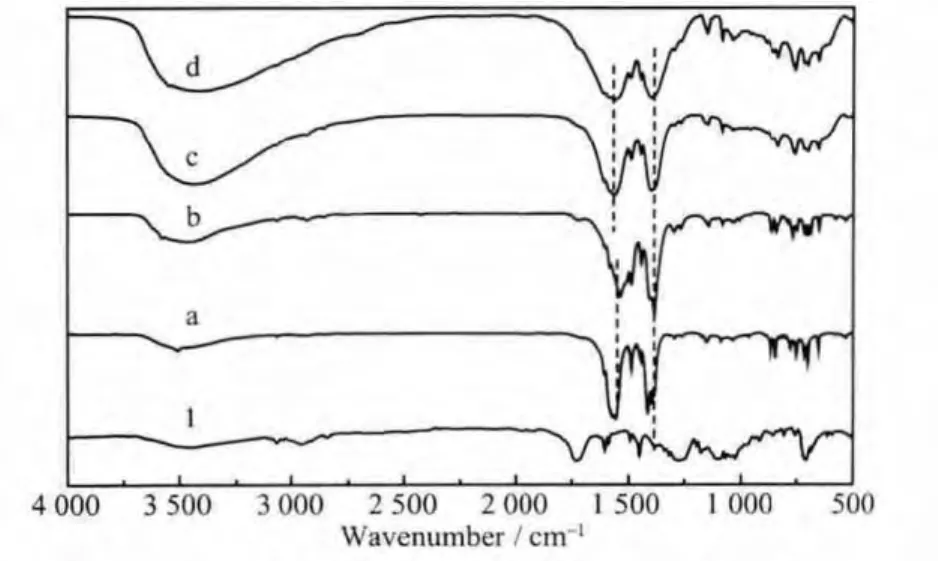
圖1 MGAE(1)及其與(a)Cd(NO3)2,(b)Pb(NO3)2,(c)ZnCl2和(d)Zn(Ac)2形成的配合物的紅外譜圖Fig.1 FT-IR spectra of the MGAE(1)and the complexes of the MGAE with Cd(NO3)2,(b)Pb(NO3)2,(c)ZnCl2,(d)Zn(Ac)2
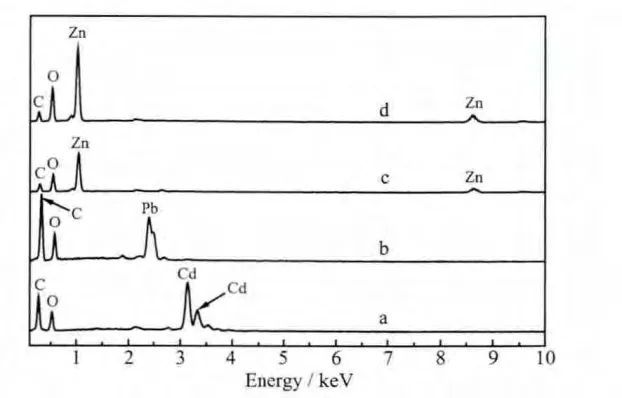
圖2 MGAE與(a)Cd(NO3)2,(b)Pb(NO3)2,(c)ZnCl2和(d)Zn(Ac)2形成的配合物的能譜圖Fig.2 Energy spectrum of the complexes of the MGAE with(a)Cd(NO3)2,(b)Pb(NO3)2,(c)ZnCl2,(d)Zn(Ac)2
2.2 結構分析
MGAE與不同的金屬鹽溶液形成的配合物的XRD衍射譜圖如圖3所示。文獻報道:合成的MOFs材料一般結晶度較差,甚至有時為非晶態物質[8],而此衍射圖譜恰恰說明了這個問題。從圖3看出,配合物的衍射圖譜均在2θ<10°的位置出現了一明顯的衍射峰,由FT-IR和XRD的分析結果可以得知,此峰可能為所得配合物的衍射峰[25]。通過比較,可得出MGAE與硝酸鎘(a)和硝酸鉛(b)形成的配合物的衍射圖譜差異較大,與氯化鋅(c)和醋酸鋅(d)形成的配合物的衍射圖譜差別不大,說明配體MGAE與不同的金屬離子配位得到的配合物在結構上具有一定的差異性。鉛離子與MGAE配體形成的配合物和鎘離子與MGAE配體形成的配合物有所不同,可能是配合鍵強度的不同、也可能是結構的不同造成的。而具有相同陽離子的2種鋅鹽與配體形成的配合物的結構基本相同。綜上,表明溶液中游離的陰離子對于配合物的形成和結構影響不明顯,而金屬陽離子對于配合物的形成和結構有明顯影響。
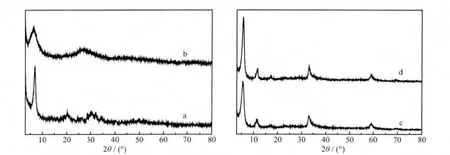
圖3 MGAE與(a)Cd(NO3)2,(b)Pb(NO3)2,(c)ZnCl2和(d)Zn(Ac)2形成的配合物的XRD圖Fig.3 XRD patterns of the complexes of MGAE with(a)Cd(NO3)2,(b)Pb(NO3)2,(c)ZnCl2,(d)Zn(Ac)2
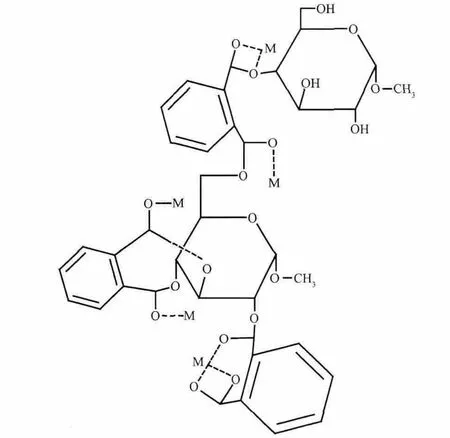
圖4 以MGAE為有機配體的部分配合物的分子結構圖Fig.4 Molecular structure of the part complex which used MGAE as organic ligands(M:metal ion)
使用ChemOffice2004建立一個以MGAE為有機配體的部分配合物的分子結構圖,如圖4所示,圖中甲基葡萄糖苷分子被3分子的鄰苯二甲酸酐酯化改性,所得到的配體中酯羰基中含有孤對電子的氧原子可直接與具有空軌道的金屬離子配位,氧原子提供電子,金屬原子提供軌道,兩者形成配位鍵,孤對電子為兩者共用。鄰苯二甲酸酐參與反應后游離出的羧基,經去質子化也可與金屬離子配位。金屬離子可通過不同的比例與配體形成配合物,主要取決于配體中孤對電子的數量、金屬離子中空軌道的數量及金屬原子和有機配體的大小所引起的空間位阻,圖示結構圖中包含配體與金屬離子以單倍、雙倍和三倍3種不同比例的配位。
2.3 TGA結果分析
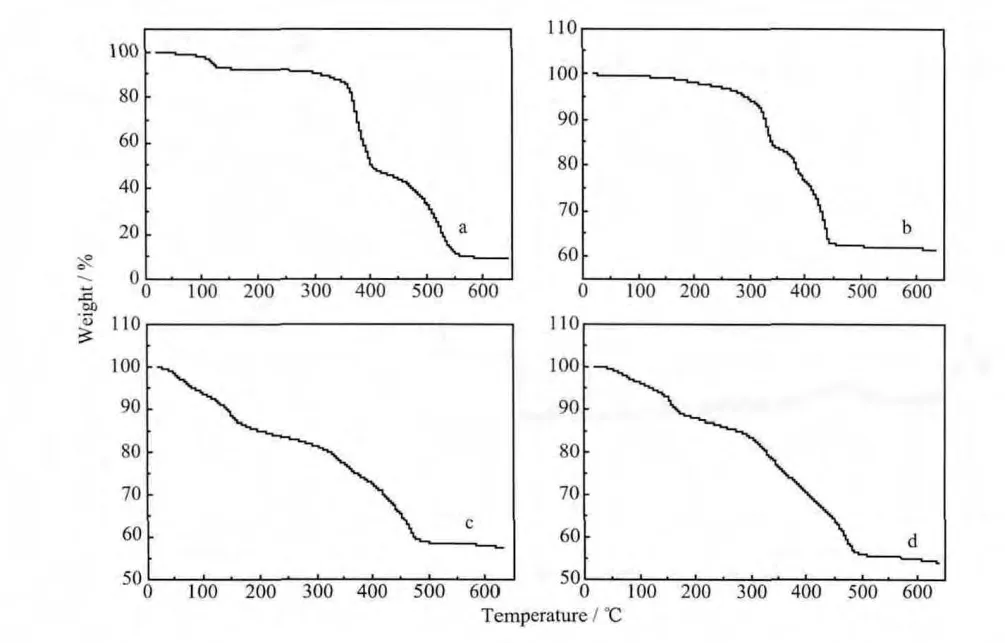
圖5 MGAE與(a)Cd(NO3)2,(b)Pb(NO3)2,(c)ZnCl2和(d)Zn(Ac)2形成的配合物的熱失重曲線圖Fig.5 TGA curves of the complexes of MGAE with(a)Cd(NO3)2,(b)Pb(NO3)2,(c)ZnCl2,(d)Zn(Ac)2
以MGAE為有機配體制備的配合物的熱失重曲線均不太規整,主要原因可能是天然產物改性后所得配體產物的結構及配體與金屬鹽溶液配位所得配合物的種類的多樣性造成的。從圖5中看出,4種配合物的全程失重都大致分為3個階段且每個階段的起止溫度相差不大,最后剩余質量大部分都為相應的鹽的氧化物。其中:第一階段失重為配合物中含有水分或是結晶水及孔道中的溶劑小分子在加熱的條件下被脫出造成的[26];第二階段失重是糖環的分解及其處于低極性化學環境的基團的分解造成的;第三階段失重是芳香環和所處極性較高的基團熱分解造成的。圖5b正如FT-IR和XRD中發現的鉛離子與MGAE配體的配位情況有別于鎘和鋅的一樣,與硝酸鉛形成的配合物的失重率為38.43%明顯低于與鎘和鋅形成的配合物,原因可能有兩方面,一方面是參與配位的有機配體比率不高;另一方面是鉛的相對質量較大。MGAE配體與氯化鋅溶液和醋酸鋅溶液形成的配合物的熱重曲線圖最為相似,更加印證了XRD的數據結果,配位過程中金屬鹽溶液中的金屬陽離子對配合物的結構和性能起決定作用。但是,總體顯示不同的金屬鹽溶液與有機配體形成的配合物的熱分解趨勢比較相似且熱穩定性較差。
2.4 FSEM結果分析
MGAE與不同金屬鹽溶液形成的配合物的掃描電鏡圖片如圖6所示。由圖可知:配合物的平均顆粒尺寸都在200 nm左右,沒有特別明確的形態[26]。與硝酸鎘形成的配合物(a)由一些不規則的片狀和少量的棒狀顆粒堆積而成,顆粒尺寸和形狀上存在較大的差異;與硝酸鉛形成的配合物(b)則由片狀的顆粒堆積而成,且由圖中可以看出一部分片狀本身存在微孔,再次印證了上面發現的鉛離子與MGAE配體的配位有別于鎘和鋅,同時這樣的形貌結構就會比配合物(a)的好一些;由圖看出與氯化鋅形成的配合物(c)和與醋酸鋅形成的配合物(d)的表面形貌在某些部分是相似的,都呈現出類似的多孔狀態,但是兩者也存在著差異,與醋酸鋅形成的配合物(d)中,多孔的形貌較少,而與氯化鋅形成的配合物(c)中,基本上都是多孔的形貌,大大增加了物質的比表面積。
2.5 BET結果分析
MGAE與不同金屬鹽溶液形成的配合物比表面積結果如表1所示。由表中看出,MGAE除了與醋酸鋅形成的配合物以外,與其他金屬鹽溶液形成的配合物的比表面積差距都不大,其中與氯化鋅形成的配合物比表面積最大,和FSEM的分析結果相吻合。但是總體看來樣品的比表面積整體偏小,可能由于(1)有機配體與金屬離子配位時無規則性致使樣品所得的孔結構不理想;(2)實驗原料采用的是生物質改性材料作為配體,樣品熱處理時溫度不能過高,過高容易導致樣品分解,而低溫會使樣品內部的客體分子不能完全去除;(3)合成的MOFs材料的孔道被溶劑分子占據[25]。實驗結果是采用在液氮的條件下通過樣品對氮氣的吸附-脫附過程來進行檢測的,由FSEM圖可以看出,有機配體與醋酸鋅形成的配合物的孔大且少,形成的晶粒尺寸大,而與醋酸鋅形成的配合物的孔多且均勻,形成的晶粒尺寸較小,可能是導致該配合物和與氯化鋅形成的配合物結構相似而比表面積差距很大的原因。

圖6 MGAE與(a)Cd(NO3)2,(b)Pd(NO3)2,(c)ZnCl2和(d)Zn(Ac)2形成的配合物的掃描電鏡圖片Fig.6 SEM images of the complexes MGAE with(a)Cd(NO3)2,(b)Pb(NO3)2,(c)ZnCl2,(d)Zn(Ac)2
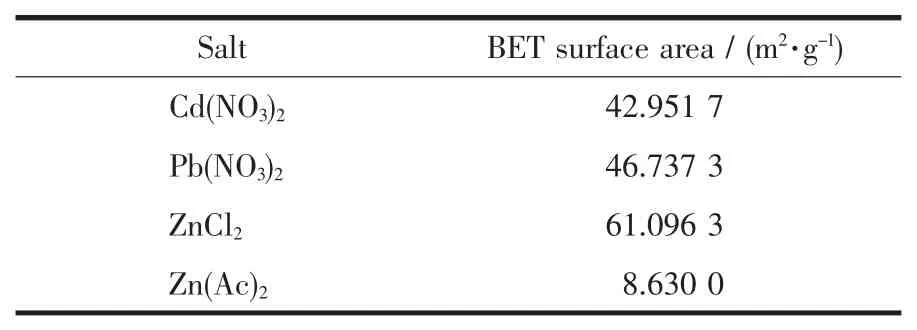
表1 MGAE-金屬配合物的比表面積Table 1 Specific surface area of the complex of the MGAE with various salts
3 結 論
本文成功的將以生物質材料——甲基葡萄糖苷為基體通過改性后得到的芳香酸酯與金屬鹽溶液配位形成金屬-有機材料,該研究為甲基葡萄糖苷探索了一種新的應用。通過研究得出:
(1)有機配體MGAE是在室溫、中性偏堿性的條件下和金屬鹽溶液進行配位的;
(2)MGAE配體與金屬鹽溶液的配位作用主要取決于羰基,且對配合物的形成和結構影響明顯的是有機配體與金屬陽離子;
(3)配合物表面多孔的形貌體現了不同金屬陽離子帶來的明顯的差別和相同金屬陽離子帶來的表面形貌的近似性;
(4)以生物質材料為原料的配合物的熱穩定性都不是很理想,且該配合物有一定的局限性,需要今后更深入的探討與研究。
[1]LONG Pei-Pei(龍沛沛),CHENG Shao-Juan(程紹娟),ZHAO Qiang(趙強),et al.Shanxi Chem.Ind.(山西化工),2008,28(6):21-25
[2]Mishra P,Mekala S,Dreisbach F,et al.Sep.Purif.Technol.,2012,94(19):124-130
[3]Rosi N L,Eckert J,Eddaoudi M,et al.Science,2003,300(5622):1127-1129
[4]Llabrés i Xamena F X,Casanova O,Galiasso Tailleur R,et al.J.Catal.,2008,255(2):220-227
[5]Llabrés i Xamena F X,Abad A,Corma A,et al.J.Catal.,2007,250(2):294-298
[6]LI Qing-Yuan(李慶遠),JI Sheng-Fu(季生福),HE Zhi-Mou(郝志謀),et al.Prog.Chem.(化學進展),2012,24(8):1506-1518
[7]Falcaro P,Normandin F,Takahashi M,et al.Adv.Mater.,2011,23(34):3901-3906
[8]WEI Wen-Fang(魏文英),FANG Jian(方鍵),KONG Hai-Ning(孔海寧),et al.Prog.Chem.(化學進展),2005,17(6):1110-1115
[9]Klimakow M,Klobes P,Rademann K,et al.Microporous Mesoporous Mater.,2012,154(15):113-118
[10]Wu G,Shi X,Fang Q R,et al.Inorg.Chem.Commun.,2003,6(4):402-404
[11]Yaghi O M,O′Keeffe M,Ockwig N W,et al.Nature,2003,423(6941):705-714
[12]Liu A Q,Huang H J,Chin L K,et al.Anal.Bioanal.Chem.,2008,391(7):2443-2452
[13]YIN Zuo-Juam(尹作娟),GAO Xiang(高翔),SUI Zhao-Lin(孫兆林),et al.Chem.Adhes.(化學與粘合),2009,31(3):61-65
[14]Yaghi O M,Li G,Li H,et al.Nature,1995,378(6558):703-706
[15]Rowsell J L C,Yaghi O M.J.Am.Chem.Soc.,2006,128(4):1304-1315
[16]Millward A R,Yaghi O M.J.Am.Chem.Soc.,2005,127(51):17998-17999
[17]Ma Y,Winnik M A,Yaneff P V,et al.J.Compos.Tech.Res.,2005,2(5):407-416
[18]LIU Li-Li(劉麗麗),ZHANG Xin(張鑫),GAO Jin-Sen(高金森),et al.Chinese.J.Catal.(催化學報),2012,33(5):833-841
[19]Fang Q R,Zhu G S,Jin Z,et al.Angew.Chem.Int.Ed.,2007,119(35):6758-6762
[20]LI Zhong-Yue(李 忠 月),LIU Kun(劉 昆),ZHANG Yun-Xing(張運星),et al.Chinese J.Inorg.Chem.(無機化學學報),2012,28(4):710-714
[21]LIU-Juan(劉 娟),LI Wei(李 偉).J.Chin.Trad.Chin.Med.Inf.(中國中醫藥咨詢),2011,3(6):270-270
[22]Mong K K T,Yen Y F,Hung W C,et al.Eur.J.Org.Chem.,2012,2012(15):3009-3017
[23]Jung W S,Chung I M,Ali M,et al.J.Asian Nat.Prod.Res.,2012,14(4):301-307
[24]Smidt E,Eckhardt K U,Lechner P,et al.Biodegradation,2005,16(1):67-79
[25]Li J,Cheng S,Zhao Q,et al.Int.J.Hydrogen Energy,2009,34(3):1377-1382
[26]Perez E V,Balkus Jr K J,Ferraris J P,et al.J.Membr.Sci.,2009,328(1):165-173
