丙酮肟水解可逆反應的進程分析和過程工藝
2014-07-24 10:30:10嚴生虎林春昕沈介發劉建武張躍
化工進展 2014年12期
嚴生虎,林春昕,沈介發,劉建武,張躍
(1 常州大學設計研究院,江蘇 常州213164;2 常州大學石油化工學院,江蘇 常州213164)
酮肟是指酮的羰基氧原子被羥氨基(=NOH)取代的一類化合物。酮肟在酸性條件下可發生水解反應,生成相應的酮和羥胺鹽,該反應是可逆反應[1-2]。然而,長期以來化學家和工程師們關注的是酮肟水解反應的逆過程,即由酮和羥胺鹽縮合生成酮肟的反應[3]。由環己酮和硫酸羥胺縮合生成環己酮肟的反應過程被發現后,由帝斯曼(DSM)等公司發展成為工業化技術,用于規模化生產己內酰胺,進而用于生產聚酰胺類聚合物尼龍-6 等[4-5],在合成材料工業中產生了深遠的影響。
隨著近年來液相氨肟化技術的發展,以TS-1為催化劑的丙酮氨肟化反應[6-7]研究逐漸見諸報道。該反應原料價廉易得,丙酮轉化率和丙酮肟選擇性最高分別達到99%和94%[7],極大地降低了丙酮肟的制備成本,有望替代傳統的羥胺鹽法工藝。同時,羥胺及其鹽作為一類重要的化工中間體,用途日益廣泛[8],傳統路線生產的羥胺鹽產品已不能滿足下游日益增長的市場需要。利用酮肟的水解反應合成羥胺鹽產品日益成為經濟可行的新路線[9]。
近年來,對酮肟水解制備羥胺鹽的可逆反應已有一些研究報道。陳林和呂俊英[9-10]研究了由丁酮肟和鹽酸反應合成鹽酸羥胺的工藝過程,羥胺鹽收率達70%以上;由于缺乏及時準確的反應進程分析方法,研究中采用最終產物稱重法進行定量測定,產物收率較低。成鳳桂、Kobayashi 等[11-12]提出了對酮肟水解反應產物進行定量分析的氧化-還原滴定法,但單一滴定分析方法無法區分羥胺濃度和酮肟濃度;Li 等[6]采用帶紫外檢測的液相色譜法(HPLC)研究了丙酮在TS-1 催化下的氨肟化反應,但該法不能檢測無紫外響應的鹽酸羥胺產物。現有方法難以準確把握酮肟水解反應進程,制約了對該反應的深入研究。作者采用自制的丙酮肟為原料,在酸性條件下進行水解反應,研究了反應進程的分析方法,創建了用于對反應體系中丙酮肟濃度、鹽酸羥胺濃度同時定量跟蹤分析的HPLC-氧化還原滴定組合測試技術。與已有報道的方法相比,本工作分析方法解決了產物稱重法無法及時跟蹤分析、誤差大的問題,解決了單一滴定法無法區分鹽酸羥胺濃度及丙酮肟濃度的問題,實現了對反應及時、準確的跟蹤分析。在此基礎上,建立了反應-蒸餾耦合工藝裝置[10],研究了丙酮肟水解可逆平衡反應[2,13]的過程工藝技術,考察了加熱溫度、蒸發速率、摩爾配比等工藝條件對鹽酸羥胺合成的影響,獲得了由丙酮肟水解制備鹽酸羥胺的優選工藝條件,為該技術的后續研究和應用奠定了基礎。
1 實驗部分
1.1 材料和裝備
丙酮肟,丙酮氨肟化法自制,GC 含量≥99.0%;38%鹽酸、硫酸高鐵銨,AR,國藥集團化學試劑公司;甲醇,色譜級,國藥集團化學試劑公司;鹽酸羥胺、硫酸羥胺,metals basis 99.99%,阿拉丁試劑公司。
反應-蒸餾耦合實驗裝置;SPD-15C 型HPLC儀,日本SHIMADZU 公司;LVS-201T 型真空泵,德國ILMVAC 公司;RCT 型磁力攪拌器,IKA 儀器公司。
1.2 實驗操作方法
丙酮肟水解制備鹽酸羥胺的可逆反應方程式如式(1)。
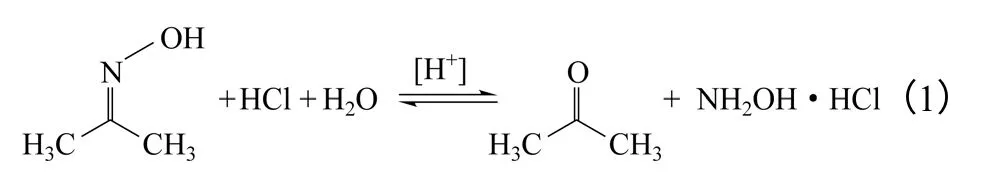
先稱量空反應瓶的質量,然后搭建反應-蒸餾耦合裝置(帶加熱器、攪拌器的反應瓶,通過氣相管路連接蒸餾管路、冷凝器及收集瓶)。在反應瓶中加入一定量的丙酮肟(記為A,mol)、一定量的鹽酸、水,加熱開始反應;生成的丙酮被及時蒸出反應體系,與蒸出的水形成混合溶液,生成的鹽酸羥胺溶解在反應區殘留液中;反應平衡向右移動,鹽酸羥胺不斷生成。反應結束后,拆卸反應裝置,稱量反應瓶及其中殘留液的質量,計算反應區殘留液質量,記為M(g);從中準確稱取出一定量樣品,記為m(g),加入容量瓶中,加入80mL 蒸餾水稀釋,再以數滴氨水中和至pH=7.0 以淬滅反應,定容至100mL,放入冰箱冷藏,用于HPLC 分析和氧化還原滴定分析,由分析結果計算反應的原料轉化率和產物收率。至反應平衡不再移動,產物組成不變后結束,產物去精制獲得鹽酸羥胺產品。
1.3 分析方法
采用本文自建的HPLC-氧化還原滴定組合分析方法對丙酮肟水解反應體系進行分析。其中,HPLC 用于測定樣品中的丙酮肟濃度Coxime(HPLC)(mol/L),外標法,分析條件為:Diamonsil C18(5μm,4.6mm×250 mm)色譜柱,流動相為甲醇︰水=7∶3(體積比,下同)的溶液,流動相流速=1mL/min,檢測波長為210nm 和265nm,進樣量為20μL。氧化還原滴定用于測定樣品中的丙酮肟和鹽酸羥胺的總濃度CTitration(mol/L),方法為:取10mL 處理后的樣品于錐形瓶中,加入5mL 25%H2SO4和10mL 250g/L 的硫酸鐵胺溶液,加熱煮沸2min 后,加入100mL 1%H3PO4水溶液,趁熱以0.01mol/L 的高錳酸鉀溶液將黃綠色溶液滴定至粉色,平行3 次,算得樣品中CTitration。
按照式(2)、式(3)計算丙酮肟水解反應的原料轉化率和產品(鹽酸羥胺)收率,鹽酸羥胺產物選擇性由收率除以轉化率計算。
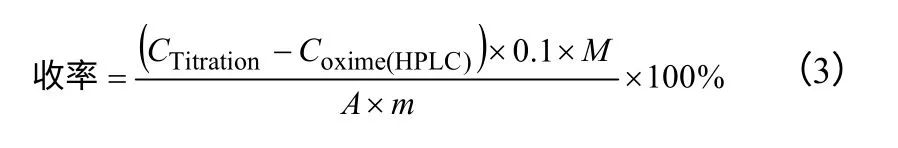
2 結果與討論
2.1 HPLC-氧化還原滴定組合測試技術
使用紫外檢測器的HPLC 可以準確測定混合樣品中的丙酮肟含量[6],但無法測出不具備紫外活性的鹽酸羥胺。鹽酸羥胺和丙酮肟均具有強還原性,可將三價鐵離子還原為二價鐵離子;同時鹽酸羥胺和丙酮肟在強酸的催化作用下均可以等摩爾轉化為NH3OH+,NH3OH+通過氧化還原滴定即可確定其含量[11,14]。丙酮肟水解反應結束后的殘留液中,存有未反應的丙酮肟和反應生成的鹽酸羥胺,單一滴定分析方法無法區分羥胺濃度和酮肟濃度。
先以氨水淬滅平衡反應,致使反應平衡不再移動,再利用HPLC 直接測定殘留液中丙酮肟的含量,避免蒸發重結晶等操作引起反應平衡繼續移動造成的誤差;然后以過量硫酸處理殘留液,將丙酮肟和鹽酸羥胺等摩爾轉化為NH3OH+,最后以氧化還原滴定測出丙酮肟和鹽酸羥胺的總量,從而計算得出鹽酸羥胺的濃度,分析方法的原理如圖1。
在上述HPLC 和氧化還原滴定的條件下,測定6 組不同濃度的丙酮肟溶液(記為Ⅰ),濃度分別為0.008mol/L、0.016mol/L、0.024mol/L、0.032mol/L、0.040mol/L、0.050mol/L;并以組合分析方法測定6組丙酮肟和鹽酸羥胺混合溶液(記為Ⅱ),濃度分別為0.34mol/L+0.06mol/L、0.28mol/L+0.12mol/L、0.22mol/L+0.18mol/L 、 0.16mol/L+0.24mol/L 、0.10mol/L+0.30mol/L、0.04mol/L+0.36mol/L,結果如表1。其中丙酮肟和鹽酸羥胺混合溶液(Ⅱ)中加入0.02mol/L 的鹽酸,以此模擬丙酮肟水解反應液中原料和產品的組成。
由表1 可見,利用本文所建立的HPLC-氧化還原滴定組合測試技術測定不同濃度丙酮肟溶液時,測定結果的平均回收率達100.69%,RSD≤1.23%(n=6),與文獻[11]以Ce4+滴定法測定單純丙酮肟樣品的回收率、RSD 水平相當。表1 中也列出了以該組合測試方法測定的混合溶液中丙酮肟濃度和鹽酸羥胺濃度,其平均回收率為99.64%,RSD≤2.04(n=6)。可見,本工作所建立的HPLC-氧化還原滴定組合測試方法具有良好的準確性和穩定性。因此,可以該分析方法為基礎,對丙酮肟水解反應的反應-蒸餾耦合工藝進行較深入的研究。
2.2 反應-蒸餾過程工藝條件的優化
2.2.1 加熱溫度對羥胺鹽合成的影響
取丙酮肟︰HCl 摩爾比為1∶2,磁力攪拌器轉速為1000r/min,反應-蒸餾時間為3h,在反應-蒸餾耦合裝置中進行丙酮肟水解反應實驗,考察了過程中不同的加熱溫度對水解反應的影響,結果如圖2。
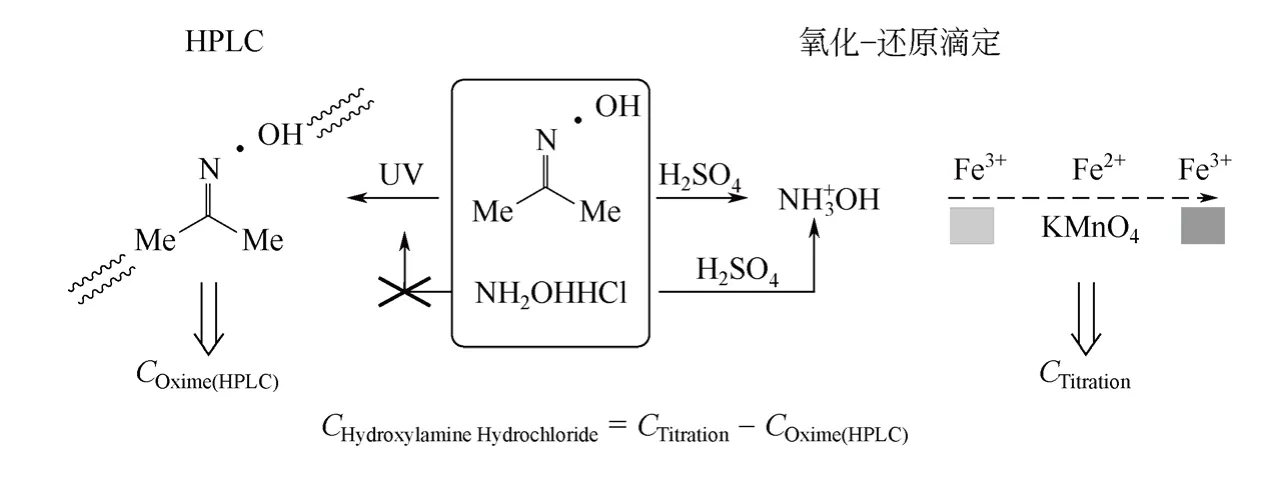
圖1 丙酮肟和鹽酸羥胺定量測定原理
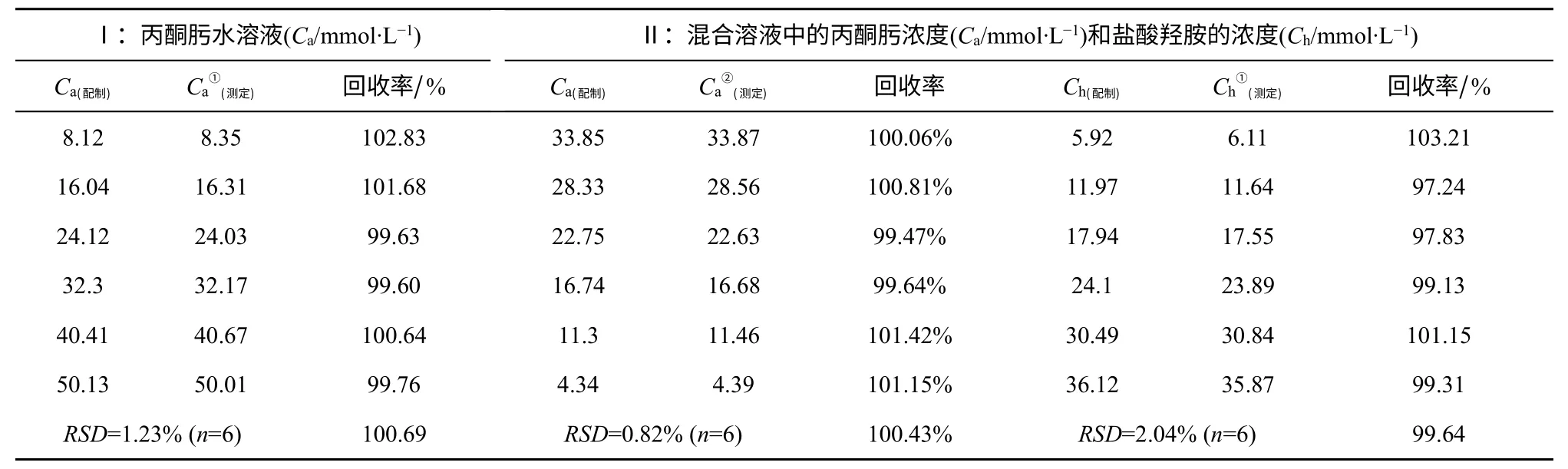
表1 丙酮肟濃度測定和混合溶液中鹽酸羥胺濃度測定結果及其回收率
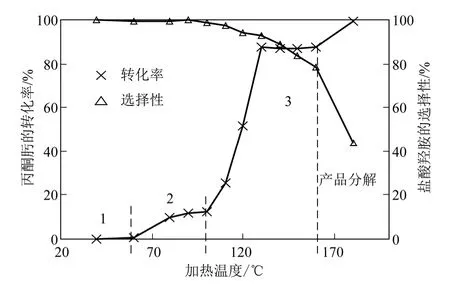
圖2 加熱溫度對丙酮肟水解反應的影響
由圖2 可見,隨著加熱溫度的上升,原料轉化率呈階梯上升趨勢,產物選擇性由緩慢降低逐漸轉為快速下降。丙酮肟的水解反應是可逆反應,1000r/min 的攪拌速率、3h 的反應-蒸餾時間足以使反應達到平衡點,反應結果表現為可逆反應的動力學平衡控制,而非傳質因素或反應時間因素限制。加熱溫度對丙酮肟水解可逆反應的原料轉化率呈現階梯式影響,包括三個階梯:階梯一(加熱溫度為40~60℃),該階段丙酮肟轉化率接近0,原因為該階段加熱溫度過低(低于丙酮的沸點),產物丙酮難以蒸發脫離反應液,致使反應平衡難以向右移動。之后,在加熱溫度60~80℃,丙酮肟轉化率通過一個明顯上升趨勢進入第二個階梯。階梯二(加熱溫度為80~100℃),該階段丙酮肟的轉化率在10%左右,且隨著加熱溫度的升高有微微上升趨勢,原因為該階段加熱溫度超過丙酮的沸點,反應-蒸餾耦合水解過程可使部分丙酮蒸發脫離反應液,實現反應平衡的移動,產物收率達到10.0%~12.4%。在階梯二的加熱溫度范圍內,水和丙酮的互溶性良好,反應-蒸餾分離出的丙酮量較少;加之反應-蒸餾同時蒸出了產物丙酮及反應物水、HCl,對正、逆反應的影響作用相當,使水解平衡未發生明顯的移動。其后,在加熱溫度100~130℃,由于超過了水的沸點,破壞了水和丙酮互溶體系,產物丙酮被大量蒸出,丙酮肟轉化率進入一個迅速上升階段,直至進入第三個階梯。文獻[10]報道在100℃加熱溫度下可獲得40%的鹽酸羥胺收率,這是因為其測算產品收率時,采用真空脫除液相組分以獲取鹽酸羥胺產物,由于過程中沒有淬滅反應,反應平衡會隨著分離過程不斷向右移動,從而出現較大的偏差,導致產物收率偏大。階梯三(加熱溫度為130~160℃),該階段丙酮肟的轉化率在89%左右,鹽酸羥胺產物收率在84%左右,原因為該階段丙酮肟轉化率已經較高,丙酮肟濃度已降低至很低的水平,可逆反應的正反應速度顯著降低,逆反應速度加快,產物丙酮一經生成即被逆反應又轉化為丙酮肟,反應-蒸餾無法再分離出更多的丙酮產物,動力學平衡點穩定不變。與以滲透汽化膜技術[15]進行酮肟水解平衡反應的工藝相比,本工作通過采用較高的加熱溫度,使產品收率提高了24%,同時避免了使用昂貴的滲透汽化膜,有利于工業化應用。考慮到過高的加熱溫度(大于160℃)易引起產品鹽酸羥胺的分解,因此選擇130℃為優選的加熱溫度。
2.2.2 蒸發速率對羥胺鹽合成的影響
取丙酮肟︰HCl 摩爾比為1∶2,磁力攪拌器轉速為1000r/min、加熱溫度為130℃,在反應-蒸餾耦合裝置中進行丙酮肟水解反應實驗,考察了0.1h、0.2h、0.5h、1h、1.5h、2h、3h、5h 對應的反應液質量變化及原料轉化率、產物選擇性的變化,并由反應液質量變化計算了不同時間段的平均蒸發速率(某時間段的平均蒸發速率為該時間段起點時刻及終點時刻的反應液質量之差除以該時間段的時長,g/min),結果如圖3。
由圖3 可見,在反應-蒸餾耦合工藝制備鹽酸羥胺剛開始的0.5h 內,蒸發速率較高,對應的丙酮肟轉化率隨時間的延長呈直線式快速增加趨勢,產物選擇性從接近100%降低至95%以上;隨著時間的延長,蒸發速率下降,丙酮肟轉化率的增速逐漸放緩,產物選擇性穩定在93%以上;當時間至2h 后,丙酮肟轉化率不再增加。蒸發過程是經典的傳熱過程,其速率取決于傳熱面積、傳熱溫差和料液的沸點。反應-蒸餾操作中料液溫度與加熱溫度接近,單位質量物料擁有的換熱面積也變化不大,因而主要影響因素是加熱溫度與沸點之差ΔT。ΔT 越大,傳熱越快,蒸發速率越快。反應剛開始的30min 內,生成的鹽酸羥胺量較少,對反應液的沸點影響較小,ΔT 保持相對恒定,反應液蒸發速率穩定在較高水平,此時反應平衡快速向右移動,產品鹽酸羥胺的不斷積累。當反應產物積累到一定程度以后,反應液沸點逐漸上升,ΔT 變小,蒸發速率下降,反應平衡的移動速度減緩,最終趨于穩定不變,達到最大的丙酮肟轉化率和鹽酸羥胺收率。由圖3 可見,丙酮肟水解反應達到最大的原料轉化率所需時間至少為2h。
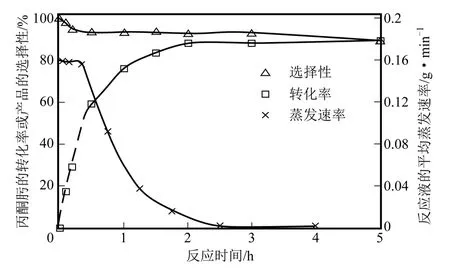
圖3 不同時間段的蒸發速率和丙酮肟水解反應結果的對應關系
2.2.3 摩爾配比對羥胺鹽合成的影響
在上述優選的工藝條件下,取磁力攪拌器轉速為1000r/min、加熱溫度為130℃、反應-蒸餾時間為3h,在反應-蒸餾耦合裝置中進行丙酮肟水解反應實驗,考察了不同丙酮肟︰HCl 摩爾比對水解反應的影響,結果如表2。
由表2 可見,隨著HCl 投料量的增加,丙酮肟轉化率和鹽酸羥胺選擇性均不斷上升,當丙酮肟︰HCl 達到1∶2 后,丙酮肟轉化率穩定在最大值89%左右,產物選擇性在92%~95%;在丙酮肟︰HCl摩爾比固定的情況下,改變鹽酸濃度對丙酮肟轉化率和產物選擇性沒有影響。這是因為鹽酸易揮發,在反應-蒸餾的過程中易被部分蒸發而脫離反應體系,使平衡反應難以繼續向右移動。增加鹽酸用量使反應體系中保留的HCl 組分濃度增大,使丙酮肟轉化率增大并趨于最大值,同時使丙酮肟發生其他不良反應的概率降低,產物選擇性增加。丙酮肟︰HCl 達到1∶2 后,丙酮肟轉化率已處于較高水平,丙酮肟濃度大幅降低,正反應速度顯著降低,逆反應速度加快,正逆反應處于動力學速度平衡狀態,丙酮肟轉化率和鹽酸羥胺選擇性趨于穩定不變。在此情況下,改變鹽酸濃度等價于改變體系中的水量,對上述動力學平衡影響微弱,對丙酮肟轉化率和產物選擇性影響很小。考慮到采用低濃度的鹽酸會產生較多酸性廢水,選擇最高可能的鹽酸濃度(質量分數38%)作為優選濃度。
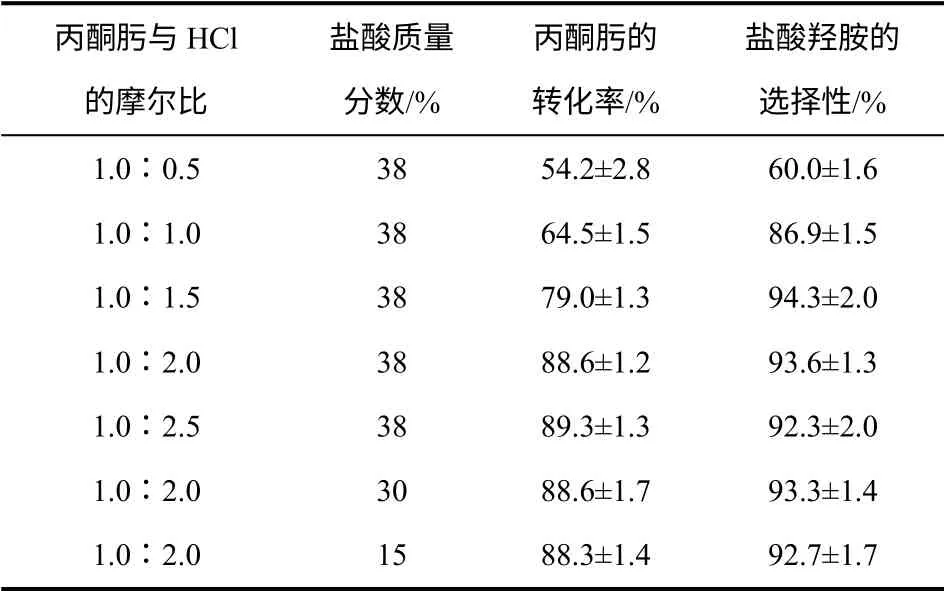
表2 丙酮肟與鹽酸的摩爾比對丙酮肟水解反應的影響
3 結 論
(1)建立了對丙酮肟水解可逆反應進程作定量分析檢測的HPLC-氧化還原滴定組合分析方法,分析結果的平均回收率達99.64%,相對標準偏差≤2.04%(n=6)。
(2)研究和優化了丙酮肟水解反應制備鹽酸羥胺的反應-蒸餾過程工藝條件,具體為:丙酮肟與鹽酸的摩爾比為1∶2,鹽酸的質量分數為38%,加熱溫度為130℃,反應時間為3h。在該過程工藝條件下,丙酮肟轉化率達到89%左右,鹽酸羥胺收率達到84%左右。
[1] Semon W L. The preparation of hydroxylamine hydrochloride and acetone oxime[J].Journal of the American Chemical Society,1923,45(1):188-190.
[2] Smith M B. March’s advanced organic chemistry :Reactions,mechanisms,and structure[M].US:Wiley,2013:1286-1287.
[3] Agne R J. Process for the production of hydroxylammonium acid sulfate:US,3066011[P].1962-11-27.
[4] Bull W C,Marhofer E G,Strickler P D,et al. Preparation of hydroxylamine:US,2827362.A[P].1953-03-18.
[5] Roffia P,Padovan M,Leofanti G,et al. Catalytic process for the manufacture of oxime:US,4794198.A[P].1988-06-08.
[6] Li Zhaohui,Chen Rizhi,Xing Weihong,et al.Continuous acetone ammoximation over TS-1 in a tubular membrane reactor[J].Industrial & Engineering Chemistry Research,2010,49(14):6309-6316.
[7] Liang X,Mi Z,Wang Y,et al.Synthesis of acetone oxime through acetone ammoximation over TS-1[J]. Reaction Kinetics and Catalysis Letters,2004,82(2):333-337.
[8] 高麗雅,檀學軍,張東升,等. 羥胺(鹽)的合成及其應用研究進展[J]. 化工進展,2012,31(9):2043-2048.
[9] 陳林. 酮肟水解制備羥胺鹽工藝[D]. 湘潭:湘潭大學,2009.
[10] 呂俊英. 固體硫酸羥胺和鹽酸羥胺的制備[D]. 杭州:浙江大學,2004.
[11] 成鳳桂,雷海. 水解-氧化還原滴定法測定鍋爐鈍化液中的丙酮肟[J]. 分析試驗室,2000,19(2):21-23.
[12] Kobayashi Y. Spectrophotometric determination of cyclohexanone oxime in sulfuric acid solution of Epsilon-Caprolactam[J]. Analytical Chemistry,1966,38(7):917-919.
[13] Jencks W P. Studies on the mechanism of oxime and semicarbazone formation1[J]. Journal of the American Chemical Society,1959,81(2):475-481.
[14] 國家質量監督檢驗檢疫總局.GB/T 6685—2007,化學試劑氯化羥胺(鹽酸羥胺)[S]. 北京:中國標準出版社,2007:2-3.
[15] 張敏.PDMS/PTFE 用于滲透汽化膜反應器的研究[D]. 北京:北京化工大學,2009.
