血清糖化清蛋白定量檢測系統(tǒng)的性能驗證
2014-09-14 06:16:48歐陽彬魏玲玲鄢盛愷
中國醫(yī)藥導報 2014年11期
歐陽彬 李 江 魏玲玲 鄢盛愷,▲
1.廣東省河源市人民醫(yī)院檢驗科,廣東河源 517000;2.中日友好醫(yī)院檢驗科,北京 100029;3.北京豪邁生物工程有限公司,北京 101318
糖化清蛋白(glycated albumin,GA)由于本身成分的單一,其測定克服了糖化血清蛋白(GSP)檢測方法學的缺陷,可以作為一個短期血糖監(jiān)測(1~2周)的可靠性指標,是糖化血紅蛋白(HbA1c)測定的有益補充[1]。近年來,GA在糖尿病患者,尤其是妊娠糖尿病患者的血糖監(jiān)測中的應(yīng)用日益廣泛。目前國內(nèi)外市場上與主流全自動生化分析儀配套使用的GA檢測試劑并不多見,而眾多國內(nèi)從事體外診斷(IVD)檢測試劑生產(chǎn)的廠家,并不生產(chǎn)與之匹配的檢測儀器。對此,臨床實驗室在使用全自動生化分析儀開放通道與開放試劑配套構(gòu)成的檢測系統(tǒng)之前,必須驗證檢測系統(tǒng)的精密度、準確度、可報告范圍、參考區(qū)間等檢測性能指標,以證明構(gòu)成的檢測系統(tǒng)性能與試劑廠商聲明的一致,符合臨床實際要求[2],而驗證這些性能指標也是目前實驗室認可(ISO 15189)過程中的重要技術(shù)要求之一。本文參照美國臨床和實驗室標準協(xié)會(CLSI)頒布的標準化文件及其他文獻方法[3-4],對北京豪邁生物工程有限公司(以下簡稱“豪邁”)自主研發(fā)生產(chǎn)的血清GA試劑盒[1]與美國貝克曼庫爾特有限公司生產(chǎn)的AU2700全自動生化分析儀(以下簡稱“AU2700生化分析儀”)組成的配套檢測系統(tǒng)進行性能驗證。
1 材料與方法
1.1 材料
采集2013年10~12月廣東省河源市人民醫(yī)院及中日友好醫(yī)院門診、住院患者和正常健康體檢者的血清樣本,混合成正常水平(GA水平11%~16%)和異常水平(GA水平>16%)的混合新鮮血清各50 mL;收集異常高濃度(GA水平 >65%)血清樣本混合成高值新鮮血清,濃度接近或高于線性檢測范圍(5 mL);采集健康體檢者新鮮血清樣本,男女各40份,每份1 mL。
1.2 試劑與儀器
豪邁GA檢測試劑盒(批號:HX006、HX005、HX004)。 豪邁 GA 校準品(批號:HX006、HX005),貝克曼庫爾特AU2700全自動生化分析儀及配套用清洗劑。
1.3 方法
按照試劑說明書中參數(shù)設(shè)定儀器參數(shù)進行實驗分析,每天測定2個水平的常規(guī)質(zhì)控品,按實驗室室內(nèi)質(zhì)控規(guī)則判斷質(zhì)控在控后,進行實驗分析。
1.3.1 準確度實驗
實驗一:使用當前校準品校準檢測體系,選取與當前校準品批號不同的另一校準品進行檢測,與試劑廠商提供的校準品說明書對照,計算相對偏差[5];實驗二:回收試驗,將不超過總體積10%的校準品溶液加入混合新鮮血清樣本中,測定計算得出回收率[6]。
1.3.2 精密度實驗
依據(jù)CLSI的EP15-A2文件中的實驗方案,驗證豪邁GA試劑盒與貝克曼庫爾特AU2700全自動生化分析儀共同構(gòu)成的檢測系統(tǒng)的精密度,每天分析1個批次樣本,每批次2個濃度,每個濃度重復(fù)測定3次,連續(xù)測定5 d,計算批內(nèi)精密度[7]。同時測定3個批次試劑,2個濃度,每個濃度重復(fù)測定3次,計算批間精密度,進而計算批內(nèi)和批間變異系數(shù)(CV),并與試劑廠家聲明的批內(nèi)CV和批間CV進行比較。
1.3.3 可報告范圍[8]
1.3.3.1 線性范圍 選擇檢測結(jié)果達到或接近廠家聲明的線性范圍上限(H)及下限(L)的新鮮血清,制成高值和低值混合樣本,低值為1號樣本,高值為6號樣本。二者以4∶1的比例混勻為2號樣本,兩者3∶2混勻為3號樣本,2∶3混勻為4號樣本,1∶4混勻為 5號樣本,混合成6份濃度不同的樣本,每個濃度的樣本重復(fù)測定2次。以預(yù)期值為±,測定均值為Y,計算回歸方程為Y=bX+a。
1.3.3.2 可報告范圍 依據(jù)試劑廠家聲明的稀釋液和稀釋倍數(shù),收集3份濃度在線性范圍內(nèi)的高值新鮮血清樣本,稀釋后分別測定,計算其稀釋回收率。
1.3.4 參考區(qū)間驗證
選擇體檢健康者,年齡18~61歲,男、女新鮮血清樣本各40份,按照CLSI頒布的C28-A2文件中的實驗方案[9]進行實驗,驗證廠商聲明的參考區(qū)間的正確性。
2 結(jié)果
2.1 準確度實驗
2.1.1 校準驗證實驗
使用批號為HX006的校準品對當前檢測系統(tǒng)進行校準,對批號為HX005的校準品進行檢測,對照被測校準品說明書,計算相對偏差,檢測值1為20.0%,檢測值2為20.2%,平均值為20.1%,低于預(yù)期值(20.6),相對偏差為2.4。GA校準品的相對偏差為2.4%。廠家的聲明是,相對偏差應(yīng)在±15%范圍內(nèi),相對偏差小于廠家聲明,符合要求。
2.1.2 回收實驗
分別將批號為 HX006 的校準品 20 μL、50 μL 和100 μL加入1000 μL正常混合新鮮血清中,校準品的標示值為20.6%,分別計算回收率。結(jié)果顯示,3個樣本的回收率分別為104.1%、107.9%和103.2%,均在試劑廠商標示的90.0%~110.0%范圍內(nèi),表明由豪邁GA試劑盒與AU2700生化分析儀組成的檢測系統(tǒng)的準確度符合要求。
2.2 精密度實驗
每天分析1個批次,2個濃度的樣本,每個濃度重復(fù)測定3次,連續(xù)測定5 d,計算批內(nèi)精密度。同時使用3個批次試劑,2個濃度,每個濃度重復(fù)測定3次,計算批間精密度。低值樣本測定均值為13.4%,驗證的CV批內(nèi)=2.42%,CV批間=2.98%,高值樣本測定均值為 24.6%,驗證的 CV批內(nèi)=1.67%,CV批間=1.89%,廠家聲明的CV批內(nèi)≤5%,CV批間≤15%,均在廠家聲明的允許范圍內(nèi)。見表2。
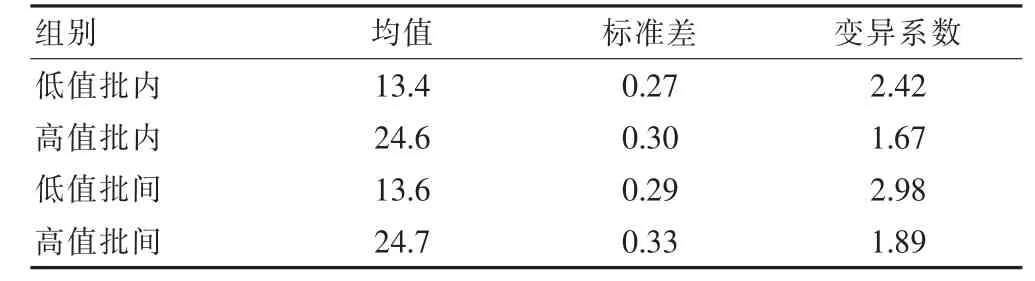
表2 精密度實驗結(jié)果(%)
2.3 臨床可報告范圍
2.3.1 線性范圍
選取測定值為3.23%的低值樣本和測定值為67.50%的高值樣本,按不同比例混合,配置成系列濃度血清,并進行雙份測定。以驗證試劑廠商聲明的GA試劑盒的線性范圍為3.20%~68.00%。見表3。
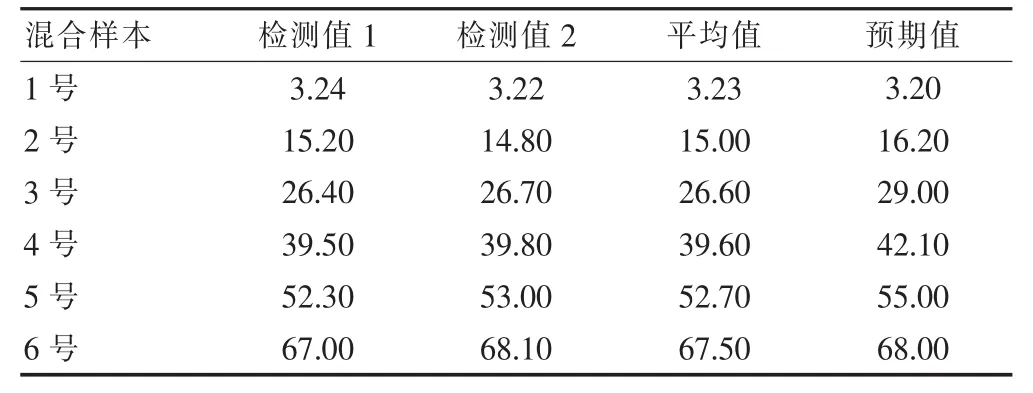
表3 線性范圍驗證(%)
2.3.2 可報告范圍
收集高值血清樣本3份,濃度在已經(jīng)驗證的GA測量范圍內(nèi),用試劑廠商推薦的生理鹽水作為稀釋液,進行最大2倍稀釋,分別重復(fù)測定3次,計算3份樣本的稀釋回收率。結(jié)果顯示,3份樣本的稀釋回收率分別為98.12%、96.86%和95.17%,均符合試劑廠商聲明的90%~110%的要求范圍,故驗證的臨床可報告范圍符合試劑廠商的3.2%~68.0%。
2.4 參考區(qū)間
40份健康成年人新鮮血清樣本測定結(jié)果顯示,所有檢測值均在試劑廠商提供的參考區(qū)間之內(nèi)(11%~16%),考慮到本實驗室的服務(wù)人群與廠商提供參考區(qū)間的人群相近,檢測方法具有可比性,故采用試劑廠商提供的參考區(qū)間進行臨床應(yīng)用是符合實際需求的。
3 討論
根據(jù)目前國內(nèi)外普遍的做法及實驗室認可的相關(guān)要求,在用于臨床檢驗之前,對于實驗室自己構(gòu)建,或是配套生化檢測系統(tǒng)應(yīng)進行評估并確認相關(guān)性能參數(shù),包括準確度、精密度、可報告范圍和參考區(qū)間,以保證所構(gòu)建的檢測系統(tǒng)符合臨床要求,證實其可以給出滿意的測定結(jié)果。利用簡便、有效的方法驗證廠家聲明的性能參數(shù),并對所用檢驗系統(tǒng)和程序進行評價,這也是實驗室質(zhì)量管理體系中的重要組成部分[10]。
本實驗采用兩種方式驗證所構(gòu)建系統(tǒng)的準確度,包括系統(tǒng)間比對試驗和系統(tǒng)內(nèi)有證參考品來驗證。本研究中,采用廠家提供定值校準品進行測定及回收實驗進行準確度驗證,用以驗證豪邁GA檢測試劑盒與AU2700生化分析儀組合構(gòu)成的檢測系統(tǒng)準確度。結(jié)果顯示,通過校準后的檢測系統(tǒng)對試劑廠商生產(chǎn)的校準品進行測定的相對偏差為2.4%。符合所聲明的相對偏差應(yīng)在±15%范圍內(nèi)。使用3個水平校準品的進行回收試驗,回收率分別為104.1%、107.9%和103.2%,均在廠商聲明的90%~110%范圍內(nèi)。通過上述兩個實驗的結(jié)果,顯示本檢測系統(tǒng)準確度符合要求。
通過精密度實驗驗證得到的批內(nèi)和批間CV均低于試劑廠商說明書標示的標準,低值樣本測定均值為 13.4%,驗證的 CV批內(nèi)=2.01%,CV批間=2.13%,高值樣本測定均值為24.6%,驗證的CV批內(nèi)=1.22%,CV批間=1.38%,均在廠家聲明的CV批內(nèi)≤5%、CV批間≤ 15%的允許范圍內(nèi),說明本檢測系統(tǒng)具有良好的精密度。
采用不同稀釋比例制成的不同濃度的系列樣本進行線性范圍驗證,發(fā)現(xiàn)試劑廠商聲明的線性范圍(3.20%~68.00%)及最大稀釋倍數(shù)實用可靠,表3結(jié)果顯示,線性回歸方程為Y=0.986X-1.005,r2=0.998,斜率在0.97~1.03之間,截距接近于0,可認為該檢測系統(tǒng)在線性范圍內(nèi)具有良好的線性檢測性能,所以可以使用生理鹽水稀釋超出線性分析范圍的樣本。按試劑說明書推薦的方法對3份高值樣本進行稀釋測定并計算回收率,顯示3份樣本的回收率分別為98.12%、96.86%和95.17%,均符合試劑廠商聲明的90%~110%范圍,說明試劑說明書所標示的線性范圍正確有效。
正確可靠的參考區(qū)間是臨床實驗室開展常規(guī)檢測工作的必要條件之一,也是臨床實驗室認可工作的基本要求。通常可以根據(jù)專業(yè)教科書或廠商說明書等資料制定參考區(qū)間,但使用不同檢測系統(tǒng)之前應(yīng)進行參考區(qū)間驗證,看是否可作為臨床實驗室實際使用的參考區(qū)間。試劑廠商提供的中國人群的GA參考區(qū)間為11%~16%,考慮到本實驗室的服務(wù)人群與豪邁廠家建立參考區(qū)間的人群相近,檢測方法具有可比性,通過驗證實驗表明,40例樣本的檢測值均在試劑廠商提供的參考區(qū)間(11%~16%)之內(nèi),所以采用此參考區(qū)間作為臨床實驗室實際應(yīng)用符合要求。
上述實驗結(jié)果顯示,由豪邁GA檢測試劑盒和與貝克曼庫爾特AU2700生化分析儀構(gòu)成的配套檢測系統(tǒng)的性能指標,包括準確度、精密度、線性范圍和臨床可報告范圍,均與豪邁公司試劑說明書聲明的一致,也說明豪邁公司提供的參考區(qū)間可以轉(zhuǎn)移使用。上述驗證實驗表明,本自建檢測系統(tǒng)的性能可以滿足臨床需求,所采用的驗證方案簡便、可行,可推廣用于臨床生化定量檢測工作中對于各種類型自建或配套檢測系統(tǒng)的性能驗證。
[1]劉常同,黃萍.糖化清蛋白在糖尿病早期診斷中的應(yīng)用價值[J].現(xiàn)代檢驗醫(yī)學雜志,2012,27(5):121-123.
[2]U.S.Department of Health and human services,centers for medicare&medicaid services.Clinical laboratory improvement amendments of 1988:final rule [S].Fed Register,2003:3704.
[3]黃小兵,劉光明,張偉堅.羅氏全自動生化分析儀自建生化檢測系統(tǒng)的性能驗證及評價[J].海南醫(yī)學,2013,24(16):2417-2419.
[4]何國堅,胡勁輝,肖慶.半胱氨酸蛋白酶抑制劑C透射比濁檢測試劑盒性能驗證[J].檢驗醫(yī)學與臨床,2013,10(23):3155-3157.
[5]張葵.定量檢測系統(tǒng)方法學性能驗證實驗的基本方法[J].臨床檢驗雜志,2009,27(5):1047-1049.
[6]王治國.臨床檢驗方法確認與性能驗證[M].北京:人民衛(wèi)生出版社,2009:242-297.
[7]Clinical and Laboratory Standards Institute.User verification of performance for precision and trueness:approved guideline second edition[S].EP15-A2(e),CLSI,2005.
[8]楊有業(yè),張秀明.臨床檢驗方法學評價[M].北京:人民衛(wèi)生出版社,2009:175-195.
[9]CLSI.C28-A2.How to define and determine reference intervals in the clinical laboratory[S].Approved Guideline-Second Edition,2000.
[10]王治國.臨床檢驗方法確認與性能驗證[M].北京:人民衛(wèi)生出版社,2009:242-297.
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數(shù)理化·中考版(2022年11期)2022-02-16 07:01:20
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2020年12期)2021-01-18 06:57:46
中學生數(shù)理化·七年級數(shù)學人教版(2020年12期)2021-01-18 06:57:46
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發(fā)明與創(chuàng)新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
