肝肺綜合征大鼠血細胞因子和肺血管巨噬細胞觀察
2014-09-20 05:49:24郭月寧王立國王夏珍
實用肝臟病雜志 2014年2期
關鍵詞:手術
郭月寧,王立國,王夏珍,張 薇
肝肺綜合征(hepatopulmonary syndrome,HPS)是指肺血管擴張、低氧血癥和一系列病理生理學變化及臨床表現。肝硬化患者最終發生肝肺綜合征的幾率高達29%,兩年半的病死率可達41%[1]。這使得找到肝肺綜合征的發病機制顯得尤為重要。但是國內外關于HPS的發病機制尚不完全明確。有研究表明,肺血管擴張是HPS最主要的病理基礎[2]。肺血管擴張并形成分流,從而引起低氧血癥,進而形成HPS是國內外比較一致的觀點。肺血管擴張是如何形成的成為了研究
的熱點。相關文獻顯示,在肝臟發生病變時,肝臟降解內毒素的功能降低,形成腸源性內毒素血癥[3]。肺臟代償性成為解毒的主要場所,內毒素刺激肺部形成肺血管巨噬細胞,肺血管巨噬細胞促進血管擴張因子的形成,引起肺部血管的擴張,進而形成肝肺綜合征。所以,有人認為內毒素血癥及肺血管巨噬細胞是促進肺血管擴張的主要根源。國內、外相關文獻顯示,采用膽總管結扎法(common bile duct ligation,CBDL)制造大鼠肝肺綜合征模型是目前較成熟的方法[4]。本實驗采用CBDL法制備肝肺綜合征模型,研究動物血內毒素水平的變化,并觀察了大鼠肺內是否出現了肺血管巨噬細胞,現報道如下。
1 材料與方法
1.1 動物 購自青島派特福德白鼠養殖專業合作社的Wistar大鼠30只,體質量約200~250 g。
1.2 建立肝肺綜合征模型 將大鼠在條件適宜的動物實驗室內飼養。隨機將動物分為三組,即假手術組(n=10)和手術組(即膽總管結扎組,CBDL組),再將CBDL組分為 CBDL 2周組(2 wk,n=10)、CBDL 5周組(5 wk,n=10)。給予4%水合氯醛1 ml.100g-1腹腔注射麻醉。假手術組大鼠沿腹白線打開腹腔后,分離膽總管,不結扎即關腹;CBDL組用4號絲線分別在近肝門處和近十二指腸端雙線結扎膽總管后關腹。在造模后2 w及5 w時處死動物,取出肝組織和肺組織,取大塊肝、肺組織放入10%中性甲醛中固定,制成蠟塊、切片,用于HE染色。
1.3 血氣分析測定 自左心室取血3~4 ml,應用血氣分析儀(美國RADISMETER公司生產的ABL700型)測定PaO2和PaCO2,以此判斷肝肺綜合征的形成。
1.4 血漿中相關指標檢測 采用凝集反應法(鱟試劑)檢測血漿內毒素(湛江博康海洋生物有限公司);采用硝酸還原酶法測定血清NO(上海研卉生物科技有限公司);采用ELISA法檢測TNF-α(上海研吉生物科技有限公司)。
1.5 腹部超聲檢查 使用加拿大超聲醫療技術有限公司生產的Sonix OP超聲診斷儀檢查膽總管擴張程度。
2 結果
2.1 血氣分析情況 在CBDL 5 wk組,動物血PaO2明顯低于假手術組(P<0.05),符合HPS特征性動脈血氣變化(表1)。
表1 各組血氣分析結果(±s)

表1 各組血氣分析結果(±s)
與假手術組比,①P<0.05
?
2.2 血漿內毒素、NO和TNF-α水平的變化 CBDL 5 wk組動物血漿內毒素、NO水平和TNF-α水平明顯高于假手術組(P<0.05,表2)。
表2 血漿內毒素、NO和TNF-α水平(±s)的變化

表2 血漿內毒素、NO和TNF-α水平(±s)的變化
與假手術組比,①P<0.05
?
2.3 膽總管擴張情況 CBDL 5 wk動物膽總管比假手術組或CBDL 2 wk組明顯擴張(圖1),而CBDL 2 wk組與假手術組比,變化不明顯(圖2)。

圖1 腹部超聲表現 CBDL 5 wk組大鼠膽總管明顯擴張
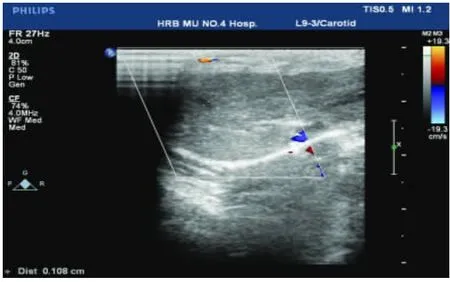
圖2 腹部超聲表現 假手術組動物膽總管無明顯擴張
2.4 肝、肺組織病理學變化 大體觀察可見CBDL 2 wk大鼠肝臟變大,淡黃色,邊緣不銳利;在膽總管結扎后5 wk,大鼠肝臟腫大,顏色加深,表面存在黃色小結節。在顯微鏡下觀察,CBDL 2 wk動物肝臟匯管區小膽管擴張,細胞呈灶狀壞死,血管周圍有大量炎細胞聚集(圖3);CBDL 5 wk組動物上述變化更加明顯,周圍結締組織出現增生并且分隔,肝細胞排列紊亂,細胞融合,細胞核固縮,部分肝細胞融解消失,形成空泡(圖4)。CBDL 5 wk組肺組織可見廣泛充血,炎性細胞浸潤,毛細血管擴張(圖5),且出現肺血管巨噬細胞(圖6)。假手術組肝肺組織無明顯變化。

圖3 肝組織病理學變化 在膽總管結扎2 wk后,肝臟炎癥細胞聚集、浸潤(HE,400×)
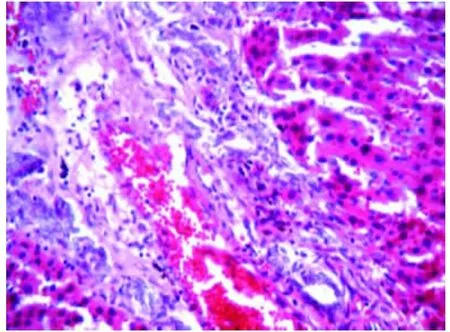
圖4 肝組織病理學變化 CBDL 5 wk組動物肝細胞排列紊亂,細胞融合,細胞核固縮,結締組織增生(HE,400×)

圖5 肺組織病理學表現 CBDL 5 wk組動物肺組織可見廣泛充血,炎性細胞浸潤,毛細血管擴張(HE,400×)

圖6 肺組織病理學表現 CBDL 5 wk組動物肺血管內出現巨噬細胞(HE,100×)
3 討論
HPS是在慢性肝病及門靜脈高壓的基礎上出現的動脈血氧合功能異常、肺血管擴張等一系列表現,在臨床上主要以進行性呼吸困難、低氧血癥為主要表現[1]。多數研究表明肝肺綜合征的本質是肝病時發生了肺血管擴張及肺動脈氧合功能降低,而引起低氧血癥[5]。
關于HPS的發病機制尚不完全明確,而肺血管擴張是引起肺通氣/灌注不匹配,出現低氧血癥的主要原因[6],這一點在國內外均較為認同。肺血管擴張在肝肺綜合征形成中起到關鍵性作用。肺血管擴張機制較為復雜。大量實驗研究表明,可能是由于肺血管內的縮血管活性因子與擴血管活性因子之間的失衡所導致的[7]。關于肺血管內擴張活性因子,如NO等的生成,有相關研究顯示是由于肝臟功能下降,消除內毒素水平降低,使得內毒素入血,進而誘導肺血管內形成巨噬細胞,肺血管巨噬細胞吞噬了血液中的異物后分泌所生成的[8]。
本實驗旨在研究內毒素及肺血管內巨噬細胞在肺血管擴張中的作用,從而更進一步研究肝肺綜合征的發生機制,為臨床診治提供更好的依據。
內毒素是導致感染性休克的最重要的生物因子。主要的作用靶細胞之一為內皮細胞,最易受到攻擊的靶器官是肺和腸[9]。內毒素本身可導致細胞自溶,由此形成腸源性內毒素血癥和腸粘膜通透性之間的惡性循環。內毒素的作用越來越受到重視[10]。最近研究證明,肝硬化大鼠細菌異位與HPS密切相關,在HPS發生時存在菌血癥和腸源性內毒素血癥,內毒素能誘導機體產生肺血管巨噬細胞,促進炎性細胞在肺血管內皮細胞粘附聚集,引起肺炎癥反應。本實驗在膽總管結扎2 wk和5 wk觀察,動物血內毒素水平隨著時間的延長而逐步升高,充分說明其在肝肺綜合征形成過程中的重要作用。
在正常大鼠肺內是沒有巨噬細胞的[11]。已有研究證明,在內毒素誘導肺急性損傷時可導致肺泡毛細血管通透性增加,促進巨噬細胞在肺內聚集,導致大量炎癥因子釋放。研究發現HPS大鼠肺內聚集大量的巨噬細胞。肺血管巨噬細胞吞噬功能增強,清除血液中的腸道菌群和內毒素,同時分泌大量的血管活性物質,包括 NO、TNF-α[12]、ET-1等,引起肺血管擴張[13]。相關研究證實,應用抑制TNF-α的制劑可以抑制腸道細菌易位,減少腸道細菌的過度生長,有效地預防HPS的發生[14]。本研究顯示,在肝肺綜合征形成過程中NO、TNF-α水平明顯升高,說明內毒素刺激肺血管巨噬細胞形成并分泌了擴血管活性因子,促進肺血管擴張,進而形成了肝肺綜合征。
CBDL大鼠可較好地復制HPS模型,并被廣泛應用于實驗研究[15]。本實驗創新點之一為在結扎膽總管時,并不對膽總管進行剪斷,減少了膽總管內膽汁對腹腔的刺激。在本研究中,動物在結扎膽總管后,隨著時間的延長,發現大鼠肝臟逐漸變大,顏色由淡黃到灰黃色,質地逐漸變硬,直至肝表面出現黃色小結節;在顯微鏡下觀察肝臟匯管區小膽管擴張,肝細胞灶狀壞死。動物血PaCO2、PaO2出現相應的病理生理學改變,NO等明顯升高,證明CBDL模型建立成功。本實驗的另一創新點為動脈血的采集。大鼠的腹主動脈在進針后易破裂,血流出后使腹腔視野變得模糊,影響肝組織的留取。經左心室取血,手術較容易,且不易破裂,采血量能夠達到血氣分析的要求。
本實驗中增加了對大鼠肝臟及結扎膽總管進行彩色多普勒超聲檢查,以便于密切關注造模情況,對于觀察模型的建造成功起到了監測作用。
肺血管擴張是HPS發病機制中出現低氧血癥的主要原因。本實驗中CBDL組在肝臟發生病理變化的同時,肺組織出現毛細血管擴張,說明肺內毛細血管擴張與肝肺綜合征的形成有關。檢測動脈血PaO2進行性下降,證明肺血管擴張是導致低氧血癥的原因。
本實驗證明,在肝肺綜合征形成過程中,內毒素起到關鍵性的重要作用,它可以刺激大鼠體內肺血管巨噬細胞的生成,進而由肺血管巨噬細胞分泌擴血管活性因子如NO等,使得肺血管出現擴張,形成低氧血癥,進而形成肝肺綜合征。但關于肺血管巨噬細胞是通過哪些信號傳導途徑引起擴血管活性因子分泌的,到現在仍不是很清楚,下一步實驗將進一步探討。
[1]Schenk P,Fuhrman V,Madl C,et al.Hepatopulmonary syndrome:prevalence and predictive value of various cut offs for arterial oxygenation and their clinical consequences.Gut,2002,51(6):853-859.
[2]Noli K,Solomon M,Golding F,et al.Prevalence of hepatopulmonary syndrome in children.Pediatrics,2008,121(3):522-527.
[3]Anderson KV.Toll signaling pathways in the innate immune response.Curr Opin Ⅰmmunol,2000,12(1):13-19.
[4]Fallon MB,Abrams GA,Luo B,et al.The role of endothelial nitric oxide synthase in the pathogenesis of a rat model of hepatopulmonary syndrome. Gastroenterology,1997,113 (2):606-614.
[5]徐希岳.肝肺綜合征.實用肝臟病雜志,1997,2(1):58-62.
[6]Mateo PA,Jose TA,Aldo TD,et al.Portopulmonary hypertension and hepatopulmonary syndrome:a clinician-oriented overview.Eur Respirat Rev,2012,21(125):223-233.
[7]倪志,孫曉寧.肝肺綜合征肺血管擴張機制的研究進展.現代生物醫學進展,2008,8(5):961-964.
[8]Parbhakar OP,Duke T,Townsend HG,et al.Depletion of pulmonary intravascular macrophages partially inhibits lipopolysaccharide-induced lung inflammation in horses.Veterin Res,2005,36(4):557-569.
[9]DeLorme Michael P,Gao Xf,Nicole DR,et al. Ⅰnflammatory effects of inhaled endotoxin-contaminated metal working fluid aerosols in rats.J Toxicol Environment Health,2003,66(1):7-24.
[10]Tilman MB,Bernhard S,Folke EB,et al.Small intestinal bacterial overgrowth in patients with cirrhosis:prevalence and relation with spontaneous bacterial peritonitis.Am J Gastroenterol,2001,96(10):2962-2967.
[11]Kawashima M,Kuwamura M,Takeya M,et al.Morphologic characteristics of pulmonary macrophages in cetaceans:particular reference to pulmonary intravascular macrophages as a newly identified type.Veterin Pathol,2004,41(6):682-686.
[12]Liu L,Liu N,Zhao ZH,et al.TNF-α neutralization improves experimental hepatopulmonary syndrome in rats.Liver Ⅰnt,2012,32(6):1018-1026.
[13]馮英梅,劉麗,劉楠,等.抗TNF-α抗體治療大鼠肝肺綜合征.基礎醫學與臨床,2012,32(1):61-65.
[14]Zhang ZH,Yang CH.Progress in investigating the pathogenesis of hepatopulmonary syndrome.Hepatobiliary Pancreat Dis Ⅰnt,2010,9(4):355-360.
[15]Vercelino R,Tieppo J,Forgiarini Junior LA,et al.Experimental models for assessment of pulmonary alterations in hepatopulmonary syndrome.J Brasil Pneumologia,2008,34(7):453-460.
猜你喜歡
環球時報(2022-12-23)2022-12-23 09:28:37
昆明醫科大學學報(2022年1期)2022-02-28 07:45:04
中老年保健(2021年11期)2021-08-22 03:13:36
昆明醫科大學學報(2021年2期)2021-03-29 07:42:46
河北畫報(2020年10期)2020-11-26 07:20:50
小學閱讀指南·低年級版(2017年1期)2017-03-13 20:07:35
中國衛生標準管理(2015年3期)2016-01-14 03:41:47
中國醫療美容(2015年1期)2015-07-12 10:06:38
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:54
西南軍醫(2014年5期)2014-04-25 07:42:48
