不同載體對負載型氧化鎢催化劑在己二酸合成反應中的結構及性能影響
2014-10-18 05:28:26張召艷祝全敬戴維林宗保寧
物理化學學報 2014年8期
張召艷 祝全敬 丁 靖 戴維林,* 宗保寧
(1復旦大學化學系,上海市分子催化和功能材料重點實驗室,上海 200433;2中國石化石油化工科學研究院,催化材料與反應工程國家重點實驗室,北京 100083)
1 引言
以WO3為活性組分的多相催化劑,由于具有酸性和氧化還原性能而廣泛應用在烯烴的選擇氧化、1-4烷烴的異構化、5氮氧化合物的選擇還原6等反應中.此外,作為一種光催化劑,WO3還有很多潛在的應用,例如能量轉換、7,8病毒失活、9有機污染物降解10,11等,由于其能帶間隙較窄(2.7-2.8 eV),在可見光下就可以表現出催化活性.12-14眾所周知,這些負載型催化劑中WO3的分散度和存在形式是影響其催化性能的主要因素.在催化劑的制備過程中,不同的制備方法、不同的制備條件、不同的載體可以得到WO3分散度、酸性和存在形態不同的催化劑,從而導致催化劑結構和性能上的差異.
迄今為止,已有很多文獻報道了WO3在不同載體上的分散情況以及存在狀態.15-19Kim等20研究了WO3在商業SiO2上的存在狀態和分散情況,通過浸漬法合成的W-Si催化劑,其表征結果說明以不同的鎢源為前驅體時,WO3在SiO2上的存在狀態也不相同.以鎢酸銨為前驅體時,在W-Si催化劑中出現了晶態WO3;而以W(η3-C3H5)4為前驅體時,沒有晶態WO3出現,室溫下鎢物種以[W12O42]12-多聚鎢物種形態存在.Zhu等21通過浸漬方法制備了WO3/ZrO2催化劑,探討了催化劑中鎢物種的存在狀態和性質對鄰苯二甲醇選擇氧化制備苯酞反應中催化性能的影響,發現當鎢物種主要以高度分散或無定形狀態存在,并且含有多聚[WO6]單元時,催化劑表現出很好的催化活性,提高焙燒溫度會導致晶態WO3的出現,使得催化活性降低.Mallesham等22通過對合成的W摻雜SnO2催化劑的研究,發現W物種可以成功地摻雜到SnO2晶格中,進而引起SnO2的晶格收縮,生成了大量的缺陷位和酸性位,提高了催化劑在甘油縮醛反應中的催化活性.Klepel等23則在堿性條件下合成了W-MCM-41介孔分子篩.他們認為,堿性條件可以有效地防止鎢物種在W-MCM-41介孔分子篩骨架外形成多聚態鎢物種和晶態WO3.紫外-可見漫反射表征表明進入分子篩骨架中的鎢物種主要以孤立或低聚態的形式存在.Koo等24成功合成了負載WO3納米顆粒的MCM-48催化劑,此催化劑對于烯烴、含硫有機物和環酮等的氧化反應具有很高的活性和選擇性.然而,該類介孔硅分子材料負載的鎢基催化劑仍然存在以下缺點:鎢物種的分散度難以控制,甚至負載量較低時,也出現細小的晶態三氧化鎢;在高溫焙燒或者氧化鎢負載量較高時,材料的介孔結構容易出現堵塞或坍塌;而焙燒溫度過低會導致鎢物種不能完全活化.因此選擇合適的載體,對于鎢物種的存在狀態和催化劑的結構形式至關重要.此外,在雙氧水體系中,鎢物種的溶脫是普遍存在的問題.因此,探索不同載體與活性物種的相互作用,使得催化劑可以多次重復利用也是研究的重點.牛新書等25采用化學共沉淀法合成了SnO2/WO3粉體,他們發現SnO2可以抑制WO3晶粒的生長,提高了其氣體靈敏度.Kamata等26報道了W/Zn/SnO2多相催化劑在烯烴及胺類等的選擇氧化反應中有非常高的活性和選擇性,并且催化劑可以多次重復使用.由此可見,WO3和SnO2之間存在較強的相互作用.近年來,WO3/SnO2復合材料作為催化劑也開始受到廣泛的關注.
己二酸(AA)是一種重要的有機化工原料,主要應用于塑料、樹脂、食品等領域,可用于制造尼龍-6,6、增塑劑、潤滑脂、聚氨酯泡沫塑料,少量用作食品的增酸劑和代替酒石酸用于發酵粉,也可作為中間體用于制造殺蟲劑和黏合劑,以及醫藥、香料等的生產.27然而,傳統己二酸的生產工藝主要是硝酸氧化法,該方法使用強氧化性的硝酸作為氧化劑,生產過程設備腐蝕嚴重,并產生大量的N2O污染物.以過氧化氫水溶液氧化環己烯合成己二酸的副產物只有水,可以實現清潔生產,具有良好的應用前景.但是,目前報道的催化劑主要是均相鎢酸催化劑,催化體系中大多使用有機溶劑、相轉移劑、助劑或有機酸配體,而這些添加劑使得反應不符合綠色工藝要求,同時產物的分離、提純和催化劑再生變得困難.28-30也有報道以硅材料或其他材料為載體,負載鎢催化劑進行己二酸的合成反應,但是這些工藝仍存在轉化率、選擇性不高或催化劑重復利用不好等問題.31-33
本文制備了一系列不同載體的負載型氧化鎢催化劑,研究了載體對于鎢物種的存在狀態的影響.首次報道了不同載體負載型氧化鎢催化劑在環氧環己烷選擇氧化制備己二酸反應中的催化性能,同時獲得了一種高活性、高選擇性和高穩定性的催化劑,為己二酸的綠色工藝開發提供了一條新穎的方法.
2 實驗部分
2.1 試劑與儀器
2.1.1 試 劑
黃鎢酸(WO3·H2O,分析純),草酸(C2H2O4·2H2O,分析純),草酸亞錫(SnC2O4,化學純),正硅酸乙酯(TEOS,分析純),無水乙醇(C2H5OH,分析純),過氧化氫(50%水溶液,分析純),濃鹽酸(37%,分析純),十二胺(DDA,化學純),P123(EO20PO70EO20,Aldrich,平均分子量5800),氫氧化鈉(NaOH,分析純).
2.1.2 儀 器
透射電鏡/場發射透射電鏡(TEM/FETEM)采用儀器型號為JEOL JEM 2011型,工作電壓為200 kV.X射線衍射(XRD)采用德國Bruker公司的D8 Advance型X射線粉末衍射儀進行催化劑樣品的物相分析,射線源采用波長為0.15405 nm的Cu Kα射線,采用Goebel鏡將發散X光束匯聚為平行光,管電壓為40 kV,管電流為40 mA,掃描速率10(°)·min-1.X射線光電子能譜(XPS)樣品測試采用Perkin-Elmer PHI 5000C ESCA System X射線光電子能譜儀,以Mg Kα射線(1253.6 eV)作為光源,分析器通能為93.9 eV,分析室的壓力<10-7Pa,X射線靶電壓為14.0 kV,功率為250 W.樣品壓片后測試,所有結合能均以污染碳(C 1s,284.6 eV)進行校正.采用SHIMADZU UV-2450型紫外-可見分光光度計進行紫外-可見漫反射光譜(UV-Vis DRS)實驗.將粉末樣品裝入樣品池中,以BaSO4為參比測定,分辨率0.1 nm.掃描范圍200-800 nm,掃描速率40 nm·min-1.激光拉曼測試在法國Jobin Yvon型共焦顯微激光拉曼光譜儀上進行,分辨率小于3 cm-1,激光能量為12.5 mW(樣品表面為2.5-3.8 mW),采用He-Ne激光器,激光波長為632.8 nm.傅里葉變換紅外(FTIR)光譜分析采用美國Nicolet公司的Avatar-360紅外光譜儀,KBr壓片,掃描范圍4000-400 cm-1,分辨率為0.9 cm-1,掃描次數32次.
2.2 催化劑的制備
SnO2:將草酸亞錫在923 K空氣中焙燒3 h(升溫速率為2 K·min-1)得到SnO2.
WO3/SnO2:采用浸漬法負載WO3.將0.5388 g H2WO4和5.4378 g H2C2O4·2H2O加入到250 mL燒杯中,然后加入215 mL去離子水,363 K下攪拌溶解,然后將2 g焙燒得到的SnO2載體分散到此溶液中,363 K下攪拌5 h,然后蒸干水分.所得固體在373 K下烘干過夜,然后在空氣氣氛的馬弗爐中以2 K·min-1的速率升溫至923 K焙燒3 h.所得粉末即為20%(質量分數,w)WO3/SnO2.
六角介孔二氧化硅(HMS)分子篩:DDA/EtOH/H2O/TEOS以摩爾比0.27:9.09:29.6:1.0投料,室溫下攪拌該混合物直至溶液澄清.在劇烈攪拌下滴加TEOS得到白色膠狀混合物.滴加完畢后保持攪拌15 min,室溫靜置老化18 h.然后過濾,用蒸餾水和乙醇洗滌后在373 K烘箱中烘干.在923 K溫度下空氣中焙燒5 h去除DDA模板劑,制得HMS分子篩.
WO3/HMS:與WO3/SnO2的負載方法一樣,僅把載體換為HMS.所得粉末即為20%WO3/HMS.
介孔分子篩(SBA-15):于313 K下,在60 mL濃HCl和312 mL水的鹽酸溶液中加入12 g P123模板劑,攪拌4 h,再加入25.6 g TEOS,攪拌24 h,然后移至水熱釜中368 K下晶化3天,取出,過濾、洗滌、干燥,于空氣中緩慢加熱到773 K,并維持6 h,得到SBA-15分子篩.
WO3/SBA-15:與WO3/SnO2的負載方法一樣,僅把載體換為SBA-15.所得粉末即為20%(w)WO3/SBA-15.
2.3 催化活性測試
在25 mL圓底燒瓶中加入0.29 g負載型鎢基催化劑,然后加入33 mmol的50%(w)雙氧水溶液,室溫攪拌,最后加入10 mmol環氧環己烷,升溫至338 K,反應2.5 h后升溫至363 K繼續反應,反應后產物通過酸堿中和滴定的方法進行定量分析.分離得率的測定采用如下方法進行.采用將反應混合物熱過濾的方法除去催化劑,在冰箱中冷卻反應混合物過夜,抽濾得到己二酸晶體,用冰水洗滌晶體二次,然后烘干后稱重,并測定熔點.催化劑重復使用方法是將反應后的催化劑離心分離,依次用去離子水和乙醇洗滌三次后在烘箱中373 K干燥3 h后備用.
3 結果與討論
3.1 催化劑的結構表征
3.1.1 XRD分析
圖1給出了純SnO2,WO3塊體及不同載體制得的負載型氧化鎢催化劑的XRD圖譜.從圖中可以看出,負載后的催化劑中都出現了歸屬于單斜WO3的(002)、(020)和(200)晶面的衍射峰,但是不同載體負載的WO3的結晶衍射峰強度不同.眾所周知,XRD衍射峰的寬度反映了樣品的結晶程度.22WO3/SnO2催化劑中 WO3的(002)、(020)和(200)晶面的衍射峰強度最弱,半峰寬較寬,說明WO3的結晶程度很低,晶粒尺寸較小.牛新書等25研究SnO2-WO3粉體時發現SnO2可以抑制WO3的晶粒生長,說明SnO2和W物種之間存在強烈的相互作用,載體SnO2可以抑制WO3的晶化和團聚.此外,在WO3/SnO2的XRD圖譜中出現了歸屬于四方晶系金紅石結構的SnO2的(110)、(101)和(211)晶面的衍射峰(JCPDS No.41-1445).當以HMS為載體時,WO3/HMS催化劑中WO3的晶態衍射峰相對較強,而WO3/SBA-15中晶態WO3衍射峰強度最強,半峰寬最小,說明負載在SBA-15上的WO3結晶程度最高,并且晶粒尺寸最大,導致其催化性能降低,與活性結果一致.
3.1.2 TEM和FETEM分析

圖1 SnO2、WO3及WO3負載在不同載體上所得催化劑的XRD圖Fig.1 XRD patterns of SnO2,WO3,and the supported tungsten oxides prepared from different supports

圖2 不同載體負載催化劑的TEM(a,b,c)及FETEM(d)照片Fig.2 TEM(a,b,c)and FETEM(d)images of catalysts with different supports
為了考察活性組分在載體表面的分散狀態,我們對不同載體的催化劑進行透射電鏡表征,結果見圖2.從圖2中可以看出,在HMS載體表面,WO3/HMS催化劑中出現了晶態WO3,并且晶粒尺寸較大(約100 nm左右);而在SBA-15表面,可以發現大量晶態WO3的聚集,說明鎢物種在SBA-15表面發生團聚,分散性很差.圖2(c)顯示了以SnO2為載體時WO3/SnO2催化劑的電鏡圖,從圖中不能明顯看見WO3物種,但是通過場發射透射電鏡圖(圖2(d))可以明顯地看見WO3物種高度分散在SnO2載體表面,并且顆粒尺寸只有2 nm左右.相對于全硅介孔分子篩HMS(1160 m2·g-1)和 SBA-15(734 m2·g-1)來說,SnO2的比表面積(13.4 m2·g-1)很小,但是W物種卻能很好地分散在其表面而沒有發生團聚,說明SnO2與W物種之間具有強烈的相互作用,是分散氧化鎢物種的良好載體.負載氧化鎢物種后,比表面積均有所降低,但氧化硅系列催化劑其比表面積還是遠遠大于WO3/SnO2催化劑.程序升溫還原分析發現,由于SnO2的可還原性,無法準確利用該技術研究鎢物種與載體的相互作用.
3.1.3 UV-Vis DRS分析
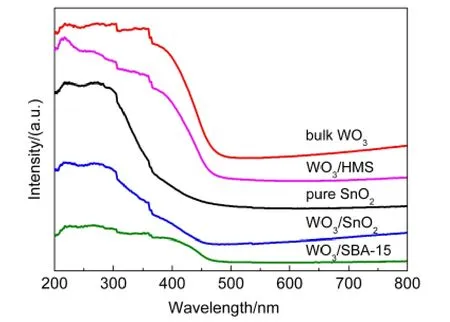
圖3 SnO2、WO3及不同載體負載催化劑的紫外-可見漫反射光譜Fig.3 UV-Vis diffuse reflectance spectra of SnO2,WO3,and catalysts with different supports
紫外-可見漫反射技術是研究過渡金屬元素離子配位情況的有效表征手段,34通過紫外-可見漫反射表征,可以更好地說明三種催化劑中鎢物種的存在狀態及分散情況.圖3顯示了不同載體催化劑的紫外-可見漫反射譜圖,其中給出了晶態WO3和SnO2載體的紫外-可見漫反射光譜作為參照.對于全硅介孔分子篩SBA-15和HMS,紫外可見漫反射圖譜中沒有檢測到明顯的特征譜峰,當負載WO3后,在230、250-350和450 nm處均出現了吸收峰,分別對應于孤立的類似于單聚鎢氧四面體[WO4]的鎢物種、低聚態的鎢物種以及晶態WO3的特征峰.35-38從WO3/HMS和WO3/SBA-15催化劑紫外可見漫反射圖譜上可以看見,在450 nm處有明顯的晶態WO3吸收峰,說明鎢物種主要以晶態WO3的形式存在,這與XRD和TEM結果相一致.但對于WO3/HMS催化劑來說,在230和250-350 nm處的吸收峰強度比WO3/SBA-15催化劑的要強,說明在WO3/HMS催化劑中單聚的鎢氧四面體[WO4]物種和低聚的鎢物種的含量要比WO3/SBA-15催化劑多,這些鎢物種主要是催化劑在焙燒過程中,鎢物種通過與載體相互作用而形成的.20對于WO3/SnO2催化劑,在230 nm和250-350 nm處也觀察到了吸收峰,說明WO3/SnO2催化劑中也存在單聚的鎢氧四面體[WO4]物種和低聚的鎢物種,此外與SnO2載體的紫外可見漫反射譜相比,在WO3/SnO2中并沒有明顯觀察到晶態WO3的吸收峰,說明以SnO2為載體時,催化劑中不存在明顯的晶態WO3,鎢物種高度分散在SnO2載體上,這一結果與FETEM觀測到的現象一致.另外,我們發現催化劑中SnO2的吸收峰向長波方向移動(與純SnO2相比),這可能是鎢物種與載體SnO2間強烈的相互作用所致.
3.1.4 Raman光譜分析
拉曼測試是一種非常靈敏的檢測表面氧化鎢顆粒的技術,尤其是對于晶態的氧化鎢非常有效.因此,Raman光譜無疑是用于檢測細小晶態WO3粒子的最有效的手段.如圖4所示,晶態WO3表現出四個特征拉曼峰,分別位于804、714、327和267 cm-1處.對于全硅分子篩HMS和SBA-15,由于自身熒光效應很強,因此其拉曼信號很弱,未出現特征拉曼峰.39對于SnO2來說,其拉曼譜峰也很弱,只在634 cm-1處可以看見歸屬于金紅石型SnO2的微弱拉曼峰.40雖然三種不同載體的負載型催化劑都表現出了晶態WO3的拉曼峰,但是峰的位置與晶態WO3拉曼峰相比都發生了一定的位移,可能是WO3物種與載體間相互作用的結果.另外,以SnO2為載體的WO3/SnO2催化劑與WO3/HMS和WO3/SBA-15催化劑相比,WO3的拉曼峰強度最弱,半峰寬最寬,說明在WO3/SnO2中WO3的結晶性能最弱,SnO2可以有效地抑制WO3的晶粒生長;而WO3/SBA-15的拉曼峰強度最強,半峰寬最窄,說明其結晶程度最高,SBA-15載體不利于WO3的分散,導致WO3的聚集,這一發現與XRD和TEM所得結果相一致.
3.1.5 XPS分析
表1列出了不同載體負載氧化鎢催化劑的表面鎢錫(硅)原子摩爾比和投料比.從表中可以看出,WO3/SnO2催化劑中表面W/Sn摩爾比要遠高于投料比,說明鎢物種主要存在于SnO2載體表面并且高度分散,而當以HMS和SBA-15為載體時,催化劑表面W/Si摩爾比反而遠小于其投料比,說明鎢物種在載體表面團聚或者進入孔道,表面分散性不好,并且,SBA-15為載體時分散性最差,這與前面的電鏡和XRD結果相一致,說明SnO2是分散鎢物種的良好載體.
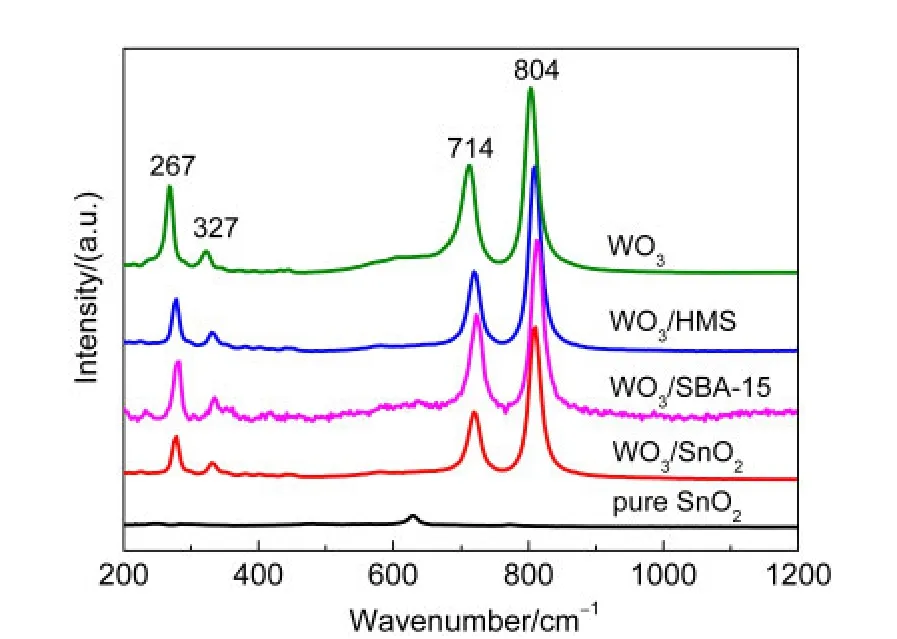
圖4 SnO2、WO3及不同載體負載催化劑的拉曼光譜Fig.4 Raman spectra of SnO2,WO3,and catalysts with different supports
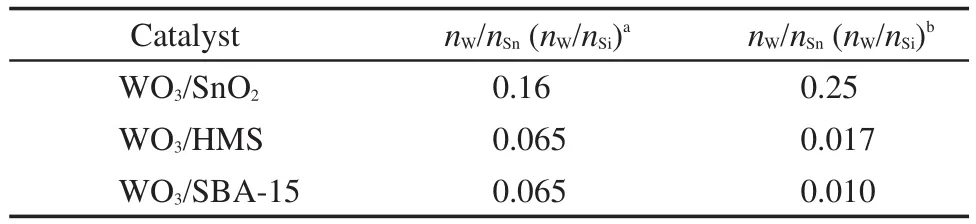
表1 不同載體負載催化劑的表面原子W/載體摩爾比Table 1 Surface W/support molar ratio of catalysts with different supports
3.1.6 FTIR分析
在合成含雜原子的負載型催化劑的研究中,紅外表征是檢測雜原子是否進入載體骨架中的一個有力證據.圖5給出WO3和不同載體催化劑的FTIR圖譜.對于全硅分子篩作為載體的催化劑中,在460、810、960及1100 cm-1處出現了硅氧振動有關的紅外吸收峰,其中460、810及1100 cm-1處的紅外譜峰分別對應于硅氧四面體骨架中的Si―O―Si的彎曲振動、對稱伸縮振動和反對稱伸縮吸收.39另外結合WO3的紅外譜圖可知,在767和825 cm-1附近出現了W―O的振動峰,因此在全硅分子篩作為載體的催化劑中,W―O的振動峰被Si―O―Si的振動所掩蓋.對于960 cm-1的歸屬卻頗有爭議,有的文獻將其歸屬于Si―OH的紅外振動峰,41但是也有研究人員認為960 cm-1處出現的譜峰是由于硅氧四面體畸變造成的骨架局部結構不對稱所致.42目前認為誘發畸變的一種原因可能是外來客體分子如摻雜的金屬離子與載體間的相互作用造成的.在含鎢的樣品中,此處的譜峰可能是在催化劑制備過程中,部分鎢物種進入到了分子篩的骨架中形成的Si―O―W所致.對于WO3/SnO2催化劑,在600-700 cm-1處的譜峰歸屬于SnO2的Sn―O―Sn振動,43同時與WO3紅外譜圖相比較,W―O的振動峰發生藍移,在750及817 cm-1左右出現了吸收峰,可能是由于WO3與SnO2之間強烈的相互作用導致的.結合前面紫外可見漫反射表征可以推測,由于鎢物種與載體之間的相互作用,部分鎢物種進入到載體晶格中,形成孤立[WO4]四面體鎢物種和低聚態的鎢物種.
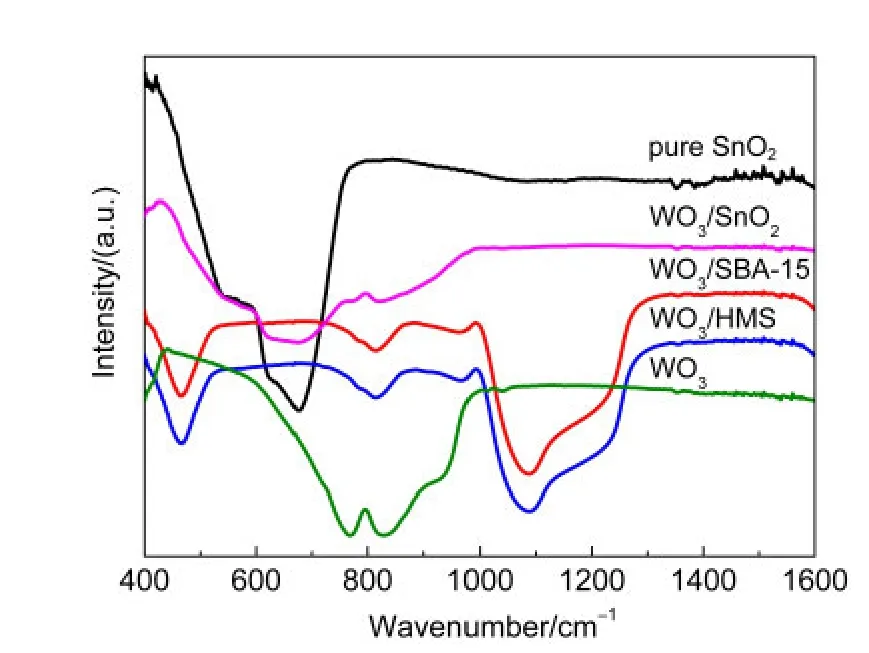
圖5 SnO2、WO3和不同載體負載催化劑的FTIR圖譜Fig.5 FTIR spectra of SnO2,WO3,and catalysts with different supports
3.2 活性測試結果
3.2.1 不同催化劑對己二酸合成的催化活性
圖6是不同催化劑的活性測試結果.從圖中可以明顯看出,己二酸的得率與催化劑的載體密切相關.以SBA-15為載體時,己二酸的得率只有53.7%,當以HMS為載體時,己二酸得率增至64.3%,但是以SnO2為載體時,催化劑表現出最好的催化活性,己二酸的得率高達96.5%.結合前面的表征結果可知,SnO2與WO3之間存在著強烈的相互作用,其可以抑制WO3晶粒的生長,并使WO3高度分散在催化劑表面,而鎢物種的高度分散能夠提供更多的活性中心,有利于催化反應的進行,這也是WO3/SnO2催化劑活性高的主要原因.通過對催化劑中O 1s分峰處理得知,WO3/SBA-15和WO3/HMS表面只存在OH氧物種,而在WO3/SnO2中還存在晶格氧,這可能也是該催化劑具有高催化活性的原因之一;并且在WO3/SnO2催化劑中還存在[WO4]四面體和低聚態的W物種,由于WO3/HMS的催化活性比WO3/SBA-15的高,而通過UV-Vis DRS表征結果可知,前者的[WO4]四面體和低聚態的W物種的含量比后者多,從而推測孤立[WO4]四面體和低聚態的W物種也可能是催化劑的活性中心,促進了反應的進行.39此外,SBA-15和HMS為載體的催化劑,活性較差的原因可能是部分鎢物種在制備過程中進入到載體孔道中,不能起到很好的催化作用,另一方面是鎢物種與載體氧化硅在反應體系中生成了鎢硅酸物種.之前已有報道稱,44與鎢同一主族的鉬的氧化物負載在以SiO2為載體的催化反應體系中,鉬物種與載體SiO2在有水的條件下,原位生成幾種鉬硅雜多酸物種,其中一些鉬硅物種的形成,降低了催化劑的催化活性.因此,這也解釋了鎢物種負載在全硅分子篩載體上催化劑活性較低的原因.另外,由于SBA-15和HMS的比表面積很大,重量很輕,因此在相同鎢用量的前提下,全硅分子篩的催化劑在無溶劑的反應體系中不能很好地分散,影響了傳質速率,從而影響其催化活性.
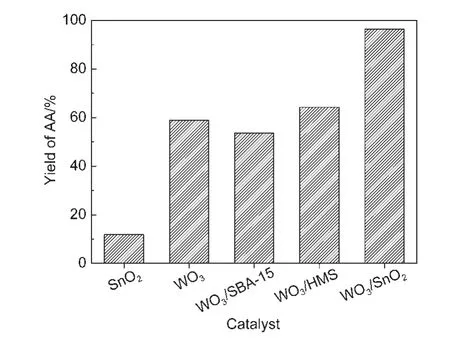
圖6 不同催化劑的催化活性Fig.6 Catalytic performance of different catalysts
3.2.2 WO3/SnO2催化劑的循環使用
為了進一步驗證鎢錫之間的強相互作用,我們對各種載體催化劑的穩定性也做了研究,催化劑的循環使用次數見圖7.從圖7可以看出,對于WO3/SnO2催化劑而言,隨著反應次數的增加,己二酸的得率緩慢地降低,催化劑在不經任何化學處理的情況下,可重復使用6次以上.第六次反應后己二酸的得率仍保持在80%以上,說明以SnO2為載體的催化劑具有良好的反應穩定性.并且催化劑經簡單焙燒后,其催化活性基本可以恢復到其初始性能,推測催化劑失活的可能原因是活性鎢物種團聚或被有機物覆蓋.而對于WO3/HMS和WO3/SBA-15催化劑,僅僅套用兩次活性就明顯降低,即使經過焙燒處理后,其活性仍然很低,說明載體與鎢物種之間相互作用很弱,催化劑穩定性很差.催化劑失活的主要原因應該是鎢物種的流失.為了驗證這一推測,我們在反應進行了4 h后,將催化劑過濾除去,濾液繼續反應.結果發現,對于WO3/SnO2催化劑,反應幾乎完全停止,然而對于WO3/SBA-15和WO3/HMS催化劑體系,反應仍在繼續進行.這一現象說明了WO3與SnO2之間存在強烈的相互作用,反應過程中鎢物種沒有溶脫,而對于WO3/SBA-15和WO3/HMS,固體催化劑過濾除去后,反應仍在繼續進行,說明溶液中存在活性均相鎢物種,催化劑失活的主要原因是鎢物種的流失.
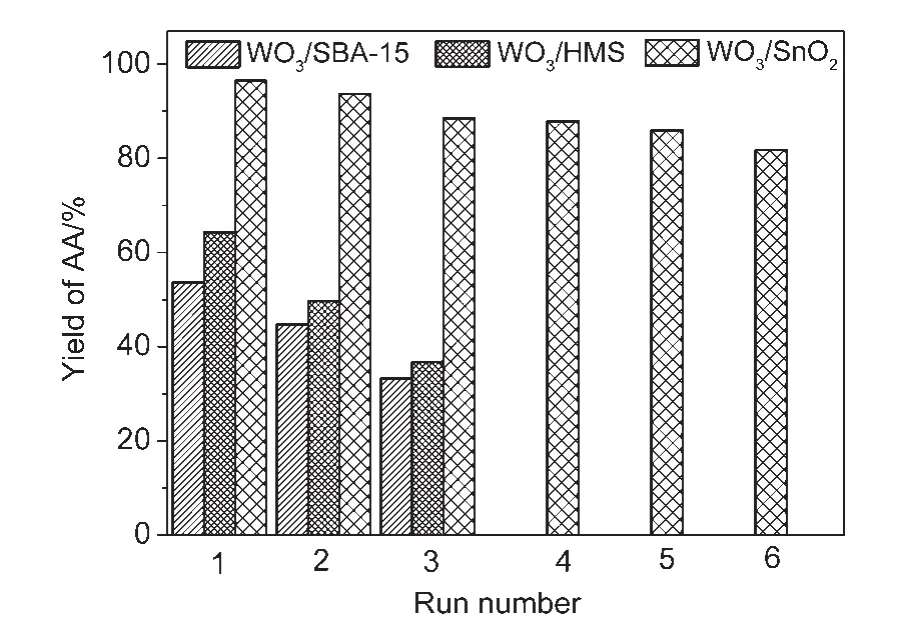
圖7 不同催化劑的穩定性測試Fig.7 Stability test of different catalysts
鑒于此,以環氧環己烷為原料,過氧化氫水溶液為氧源,具有高活性高穩定性的WO3/SnO2作為催化劑的己二酸綠色合成工藝,具有良好的實際應用前景.該路線原料易得,生產過程潔凈,無有機溶劑和相轉移催化劑,不產生氮氧化物等污染物,如大規模工業化可以改變目前工業己二酸生產工藝污染嚴重的狀況,并且多相催化劑分離簡單,產物易提純,為己二酸的新型綠色工藝開發提供了一條新穎的方法.
4 結論
通過簡單的浸漬法合成了不同載體的鎢基催化劑,并應用到己二酸合成反應中.催化活性結果顯示,當以SnO2為載體時,鎢基催化劑的催化活性最好,己二酸的得率高達96%以上.而以SBA-15和HMS為載體時,催化劑的催化活性較差,說明載體對催化劑的催化性能有重要的影響.通過表征結果可得知,以SnO2為載體的WO3/SnO2催化劑中氧化鎢物種結晶程度較低,粒徑很小,并且高度分散在催化劑表面,這是催化劑具有高活性的主要原因.經過穩定性實驗發現,WO3/SnO2可以重復使用6次,并且己二酸的得率仍保持在80%以上,說明鎢物種與SnO2載體之間存在強烈的相互作用,使催化劑具有高穩定性,為己二酸的新型綠色合成工藝開發提供了一條新途徑.
(1)Yang,X.L.;Yin,A.Y.;Dai,W.L.;Fan,K.N.Acta Phys.-Chim.Sin.2011,27(1),177.[楊新麗,尹安遠,戴維林,范康年.物理化學學報,2011,27(1),177.]doi:10.3866/PKU.WHXB20110105
(3)Yang,X.L.;Dai,W.L.;Chen,H.;Cao,Y.;Li,H.X.;He,H.Y.;Fan,K.N.J.Catal.2005,229,259.
(4)Sels,B.F.;Devos,D.E.;Jacobs,P.A.Angew.Chem.Int.Edit.2005,44(2),310.
(5)Wilson,R.D.;Barton,D.G.;Baertsch,C.D.;Iglesia,E.J.Catal.2000,194(2),175.doi:10.1006/jcat.2000.2942
(6)Engweiler,J.;Harf,J.;Baiker,A.J.Catal.1996,159(2),259.doi:10.1006/jcat.1996.0087
(7)Sivula,K.;Formal,F.L.;Gr?tzel,M.Chem.Mater.2009,21,2862.doi:10.1021/cm900565a
(8)Ham,D.J.;Phuruangrat,A.;Thongtem,S.;Lee,J.S.Chem.Eng.J.2010,165,365.doi:10.1016/j.cej.2010.09.003
(9)Takehara,K.;Yamazaki,K.;Miyazaki,M.;Yamada,Y.;Ruenphet,S.;Jahangir,A.;Shoham,D.;Okamura,M.;Nakamura,M.Virus Res.2010,151,102.doi:10.1016/j.virusres.2010.03.006
(10)Abe,R.;Takami,H.;Murakami,N.;Ohtani,B.J.Am.Chem.Soc.2008,130,7780.doi:10.1021/ja800835q
(11)Qamar,M.;Gondal,M.A.;Yamani,Z.H.Catal.Commun.2010,11,768.doi:10.1016/j.catcom.2010.02.012
(12)Morales,W.;Cason,M.;Aina,O.;Tacconi,N.R.;Rajeshwar,K.J.Am.Chem.Soc.2008,130,6318.doi:10.1021/ja8012402
(13)Huang,L.Y.;Xu,H.;Li,Y.P.;Li,H.M.;Cheng,X.N.;Xia,J.X.;Xu,Y.G.;Cai,G.B.Dalton Trans.2013,42,8606.doi:10.1039/c3dt00115f
(14)Li,F.B.;Gu,G.B.;Li,X.J.;Wan,H.F.Acta Phys.-Chim.Sin.2000,16(11),997.[李芳柏,古國榜,李新軍,萬洪富.物理化學學報,2000,16(11),997.]doi:10.3866/PKU.WHXB20001108
(15)Horsley,J.A.;Wachs,I.E.;Brown,J.M.;Via,G.H.;Hardcastle,F.D.J.Phys.Chem.1987,91(15),4014.doi:10.1021/j100299a018
(16)Engweiler,J.;Harf,J.;Baiker,A.J.Catal.1996,159(2),259.doi:10.1006/jcat.1996.0087
(17)Hilbrig,F.;G?bel,H.E.;Kn?zinger,H.;Schmelz,H.;Lengeler,B.J.Phys.Chem.1991,95(18),6973.doi:10.1021/j100171a046
(18)Colque,S.;Payen,E.;Grange,P.J.Mater.Chem.1994,4(8),1343.doi:10.1039/jm9940401343
(19)Kim,D.S.;Ostromecki,M.;Wachs,I.E.J.Mol.Catal.A:Chem.1996,106(1-2),93.doi:10.1016/1381-1169(95)00186-7
(20)Kim,D.S.;Ostromecki,M.;Wachs,I.E.;Kohler,S.D.;Ekerdt,J.G.Catal.Lett.1995,33(3-4),209.doi:10.1007/BF00814225
(21)Zhu,Q.J.;Chu,X.F.;Zhang,Z.Y.;Dai,W.L.;Fan,K.N.Appl.Catal.A:Gen.2012,435-436,141.
(22)Mallesham,B.;Sudarsanam,P.;Raju,G.;Reddy,B.M.Green Chem.2013,15(2),478.doi:10.1039/c2gc36152c
(23)Klepel,O.;B?hlmann,W.;Ivanov,E.B.;Riede,V.;Papp,H.Microporous Mesoporous Mat.2004,76(1-3),105.doi:10.1016/j.micromeso.2004.07.038
(24)Koo,D.H.;Kim,M.;Chang,S.Org.Lett.2005,7(22),5015.doi:10.1021/ol052019i
(25)Niu,X.S.;Liu,Y.L.;Hu,P.;Xu,J.Q.Electron.Comp.Mater.2002,21(1),10.[牛新書,劉艷麗,胡 平,徐甲強.電子元件與材料,2002,21(1),10.]
(26)Kamata,K.;Yonehara,K.;Sumida,Y.;Hirata,K.;Nojima,S.;Mizuno,N.Angew.Chem.Int.Edit.2011,50(50),12062.doi:10.1002/anie.v50.50
(27)Wang,J.M.Chem.Technol.Market 2010,33(11),1.[汪家銘.化工科技市場,2010,33(11),1.]
(28)Penate,I.Q.;Lesage,G.;Cognet,P.;Poux,M.Chem.Eng.J.2012,200-202,357.
(29)Wei,L.;Chen,M.;Liu,N.;Wang,S.J.;Wang,J.F.J.Dalian Polytech.University 2010,29(3),216.[魏 莉,陳 梅,劉娜,王少君,王吉峰.大連工業大學學報,2010,29(3),216.]
(30)Jiang,H.;Gong,H.;Yang,Z.H.;Zhang,X.T.;Sun,Z.L.;Kinet,R.Catal.Lett.2002,75(2),315.doi:10.1023/A:1015207214720
(31)Cheng,C.Y.;Lin,K.J.;Prasad,M.R.;Fu,S.J.;Chang,S.Y.;Shyu,S.G.;Sheu,H.S.;Chen,C.H.;Chuang,C.H.;Lin,M.T.Catal.Commun.2007,No.8,1060.
(32)Bohstrom,Z.;Lattes,I.R.;Holmberg,K.Green Chem.2010,12,1861.doi:10.1039/c0gc00032a
(33)Sheng,X.L.;Zhou,Y.M.;Zhang,Y.W.;Duan,Y.Z.;Xue,M.W.Catal.Lett.2012,142,360.doi:10.1007/s10562-012-0769-5
(34)Stein,A.;Fendorf,M.;Jarvie,T.P.;Mueller,K.T.;Benesi,A.J.;Mallouk,T.E.Chem.Mater.1995,7(2),304.doi:10.1021/cm00050a012
(35)Briot,E.;Piquemat,J.Y.;Vennat,M.;Brégeault,J.M.;Chottard,G.;Manoli,J.M.J.Mater.Chem.2000,10(4),953.doi:10.1039/a908428b
(36)Klepel,O.;B?hlmann,W.;Ivanov,E.B.;Riede,V.;Papp,H.Microporous Mesoporous Mat.2004,76(1-3),105.doi:10.1016/j.micromeso.2004.07.038
(37)Weber,R.S.J.Catal.1995,151(2),470.doi:10.1006/jcat.1995.1052
(38)Iglesia,E.;Barton,D.G.;Soled,S.L.;Miseo,S.;Baumgartner,J.E.;Gates,W.E.;Fuentes,G.A.;Meitzner,G.D.Stud.Surf.Sci.Catal.1996,101,533.doi:10.1016/S0167-2991(96)80264-3
(39)Yang,X.L.;Dai,W.L.;Chen,H.;Xu,J.H.;Cao,Y.;Li,H.X.;Fan,K.N.Appl.Catal.A:Gen.2005,283,1.doi:10.1016/j.apcata.2004.12.029
(40)Ansari,S.G.;Dar,M.A.;Dhage,M.S.;Kim,Y.S.;Ansari,Z.A.;Al-Hajry,A.;Shin,H.S.Rev.Sci.Instrum.2009,80(4),045112-1.doi:10.1063/1.3115222
(41)Chen,C.Y.;Li,H.X.;Davis,M.E.Micropor.Mater.1993,2(1),17.doi:10.1016/0927-6513(93)80058-3
(42)Fu,Z.H.;Yin,D.L.;Xie,Q.J.;Zhao,W.;Lv,A.;Yin,D.H.;Xu,Y.Z.;Zhang,L.X.J.Mol.Catal.A:Chem.2004,208(1-2),159.doi:10.1016/S1381-1169(03)00508-9
(43)Zhu,J.J.;Lu,Z.H.;Aruna,S.T.;Aurbach,D.;Aharon,G.Chem.Mater.2000,12,2557.doi:10.1021/cm990683l
(44)Kotbagi,T.V.;Biradar,A.V.;Umbarkar,S.B.;Dongare,M.K.ChemCatChem 2013,5,1531.doi:10.1002/cctc.v5.6
猜你喜歡
課堂內外·初中版(科學少年)(2025年1期)2025-02-28 00:00:00
課堂內外·初中版(科學少年)(2025年2期)2025-02-28 00:00:00
英語世界(2023年10期)2023-11-17 09:18:18
科學大眾(中學)(2019年3期)2019-05-17 10:04:30
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
汽車觀察(2018年10期)2018-11-06 07:05:26
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
