高效液相色譜-蒸發光散射檢測器法測定氟比洛芬酯注射液中大豆油含量
2014-11-08 08:35:40張勉,李超
中國藥業 2014年7期
張 勉,李 超
(重慶市食品藥品檢驗所,重慶 401121)
氟比洛芬酯注射液是以脂微球為藥物載體的非甾體類鎮痛劑。藥物進入體內靶向分布到創傷及腫瘤部位后,氟比洛芬酯從脂微球中釋放出來,在羧基酯酶作用下迅速水解生成氟比洛芬,通過氟比洛芬抑制前列腺素的合成而發揮鎮痛作用。大豆油是制備乳劑的重要原輔料,其含量直接影響乳劑的成型質量,在氟比洛芬酯注射液中大豆油作為氟比洛芬酯的載體,有利于氟比洛芬酯達到靶向部位,更好地吸收。但由于大豆油為長鏈甘油三酸酯,紫外特征不強,常規的紫外檢測器無法完成對其的測定。國內僅有一個廠家生產該品種,現行質量標準[1]中未對大豆油進行含量控制。筆者采用高效液相色譜法分離,利用蒸發光散射檢測器測定氟比洛芬酯注射液中大豆油的含量,有效地解決了大豆油在該注射劑中的檢測問題,方法準確,操作簡便。
1 儀器與試藥
Waters e2695型高效液相色譜儀。大豆油對照品(中國藥品生物制品檢定所,批號為100277-201103);大豆油原料(重慶藥友制藥有限責任公司,批號為R1105067);氟比洛芬酯注射液(北京泰德制藥股份有限公司,批號分別為5091N,5012B,5022B);正己烷與異丙醇均為色譜純,冰醋酸為分析純。
2 方法與結果
2.1 色譜條件與系統適用性試驗
檢測器:Waters 2424型蒸發光檢測器(霧化氣:空氣,霧化壓力:35.0 psi,漂移管溫度:70℃);色譜柱:菲羅門 Luna Silica色譜柱(250 mm ×4.6 mm,5 μm);流動相:正己烷 - 異丙醇 - 冰醋酸(99.4 ∶0.5 ∶0.1);流速:1.0 mL/min;柱溫:室溫。進樣量:10 μL。配制大豆油與油酸的混合溶液,按擬訂方法測定,兩者能較好地分離,見圖1。
2.2 溶液制備
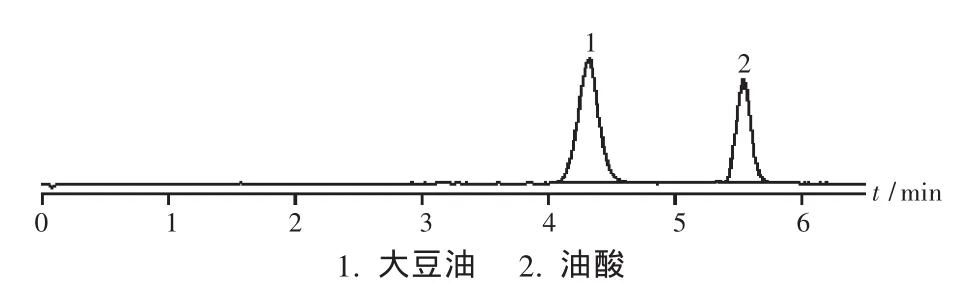
圖1 高效液相色譜圖
取大豆油對照品0.062 11 g,置25 mL容量瓶中,用正己烷-異丙醇(1∶1)的混合溶液溶解并稀釋至刻度,搖勻,分別精密量取 1.0,3.0,5.0 mL,分別置 10 mL 容量瓶中,加混合溶液稀釋至刻度,搖勻,作為對照品溶液。精密量取樣品1 mL,置25 mL容量瓶中,用混合溶液溶解并稀釋至刻度,搖勻,分別精密取3 mL,置25mL容量瓶中,加混合溶液稀釋至刻度,搖勻,即得供試品溶液。
2.3 大豆油原料的標定
取大豆油原料0.543 2 g,置100 mL容量瓶中,加混合溶液溶解并稀釋至刻度,搖勻,精密量取5.0 mL,置50 mL容量瓶中,加混合溶液稀釋至刻度,搖勻,依法測定,測得大豆油含量為 100.0% 。
2.4 方法學考察
線性關系考察:取大豆油原料0.853 6 g,置100 mL容量瓶中,加混合溶液溶解并稀釋至刻度,搖勻,分別精密量取1,2,4,5,6,8,10 mL,分別置50 mL容量瓶中,加混合溶液稀釋至刻度,搖勻,依法測定,得回歸方程 Y=0.001 7 X+115.7(r=0.998)。結果表明,大豆油質量濃度在 1.707~17.072 μg/mL范圍內與峰面積呈良好線性關系。
精密度試驗:取同一質量濃度對照品溶液,連續進樣6次。結果的 RSD為0.57%(n=6),表明儀器精密度良好。
重復性試驗:取樣品(批號為5091N)各1.0 mL,共6份,分別置25 mL容量瓶中,加混合溶液溶解并稀釋至刻度,搖勻,分別精密量取3.0 mL,置25 mL容量瓶中,加混合溶液稀釋至刻度,搖勻,依法測定。結果樣品平均含量為10.5 g/mL,RSD為2.4%,表明方法重復性良好。
加樣回收試驗:取大豆油原料0.543 2 g,置100 mL容量瓶中,加混合溶液溶解并稀釋至刻度,搖勻,作為對照品貯備液。取樣品(批號為5091N)各 1.0 mL,共9份,分別置25 mL容量瓶中,加混合溶液溶解并稀釋至刻度,搖勻,分別精密量取3.0 mL,置 50 mL 容量瓶中,分別加入對照品貯備液 2.0,2.5,3.0 mL,各3份,加混合溶液稀釋至刻度,搖勻,依法測定。結果見表1。
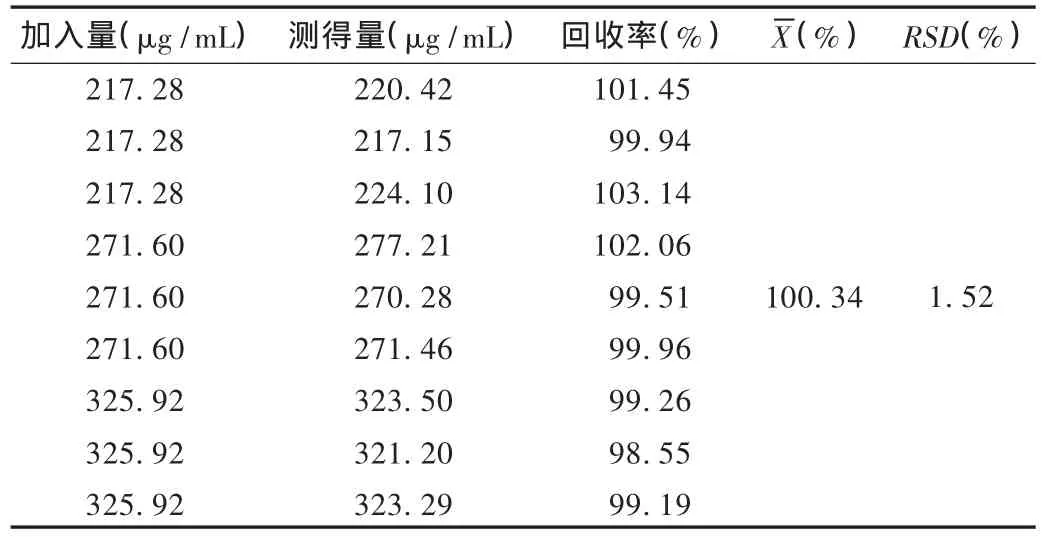
表1 大豆油加樣回收試驗結果(n=9)
檢測限及定量限:以 S/N為3計算,大豆油的檢測限為0.050 7 μg。以 S/N 為 10 計算,大豆油的定量限為 0.169 μg。
專屬性試驗:取樣品1 mL,置錐形瓶中,加0.1 mol/L鹽酸溶液2 mL,加熱至沸,放冷,加0.1 mol/L氫氧化鈉溶液2 mL,加混合溶液稀釋至25 mL,搖勻,取3.0 mL,置25 mL容量瓶中,加混合溶液稀釋至刻度,搖勻,作為酸破壞溶液。取樣品1 mL,置錐形瓶中,加0.1 mol/L氫氧化鈉溶液2 mL,加熱至沸,放冷,加0.1 mol/L鹽酸溶液2 mL,加混合溶液稀釋至25 mL,搖勻,取3.0 mL,置25 mL容量瓶中,加混合溶液稀釋至刻度,搖勻,作為堿破壞溶液。取樣品1 mL,置錐形瓶中,加30%過氧化氫溶液2 mL,加熱至沸,放冷,加混合溶液稀釋至25 mL,搖勻,取3.0 mL,置25 mL容量瓶中,加混合溶液稀釋至刻度,搖勻,作為氧化破壞溶液。取樣品1 mL,置錐形瓶中,加水2 mL,加熱至沸,放冷,加混合溶液稀釋至25 mL,搖勻,取3.0 mL,置25 mL容量瓶中,加混合溶液稀釋至刻度,搖勻,作為高溫破壞溶液。取樣品置日光下放置48 h后,取1 mL置25 mL容量瓶中,加混合溶液稀釋至刻度,搖勻,取3.0 mL,置25 mL容量瓶中,加混合溶液稀釋至刻度,搖勻,作為光破壞溶液。取上述破壞后的供試品溶液,按擬訂色譜條件分別注入高效液相色譜儀,記錄色譜圖,見圖3。可見,本品經破壞后產生的降解產物均能與大豆油有效分離(分離度大于1.5),表明上述色譜條件可用于大豆油含量的測定。
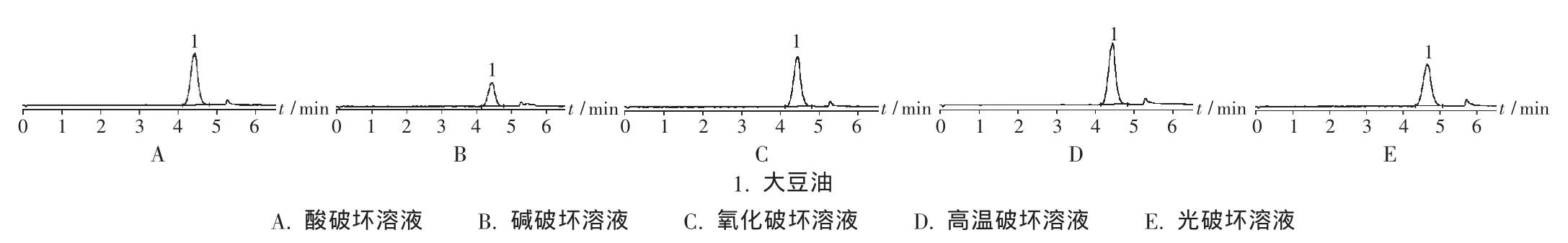
圖3 高效液相色譜圖
2.5 樣品含量測定
按擬訂方法測定3批氟比洛芬酯注射液中大豆油的含量。結果批號為5091N,5012B,5022B的大3批氟比洛芬注射液中豆油含量分別為 10.8,10.7,10.5 g/mL。
3 討論
參照脂肪乳注射液(C14-24)質量標準[2]中大豆油含量測定項下方法,流動相為正己烷 -異丙醇 -冰醋酸(98.9∶1∶0.1),大豆油和油酸的分離較差,達不到要求。因此將流動相調整為正己烷-異丙醇 - 冰醋酸(99.4 ∶0.5 ∶0.1),大豆油和油酸的分離度大于 2。
參照脂肪乳注射液(C14-24)質量標準[2]中大豆油含量測定項下方法,在供試品溶液配制時先用正己烷-異丙醇(1∶1)的混合溶液溶解,再用流動相稀釋,供試品溶液即發生乳渾。經考察,在第二步仍用上述混合溶液稀釋,溶液澄清。因此試驗中所用溶劑均為正己烷-異丙醇(1∶1)混合溶液。
在結果計算時,峰面積和質量濃度計算的線性良好,所以未使用峰面積和質量濃度的對數進行計算。
[1]YBH15412004,國家食品藥品監督管理局標準(試行)[S].
[2]YBH13472008,國家食品藥品監督管理局標準[S].
