氣相色譜法測(cè)定苦參蒼術(shù)口服液中三氯甲烷殘留量
2014-11-23 03:55:08劉自揚(yáng)萬(wàn)仁玲段文龍
中國(guó)獸藥雜志 2014年2期
范 強(qiáng),劉自揚(yáng),萬(wàn)仁玲,段文龍
(中國(guó)獸醫(yī)藥品監(jiān)察所,北京100081)
苦參蒼術(shù)口服液由苦參和蒼術(shù)兩味中藥組成,具有清熱、燥濕、止痢之功效,主治雞大腸桿菌病,其在制劑制備過程中使用三氯甲烷提取苦參總生物堿。三氯甲烷是一種強(qiáng)揮發(fā)性物質(zhì),高劑量的三氯甲烷能抑制中樞神經(jīng),并可造成肝腎損害及腫瘤[1]。《中華人民共和國(guó)獸藥典》二○一○年版規(guī)定,三氯甲烷屬第二類殘留溶劑,應(yīng)限制使用,限度為0.006%[2],控制其殘留量對(duì)保證獸藥質(zhì)量和用藥安全具有重要意義。因此,本研究按照《中國(guó)獸藥典》殘留溶劑測(cè)定法的要求,在參考相關(guān)文獻(xiàn)基礎(chǔ)上[3-5],建立了苦參蒼術(shù)口服液中三氯甲烷殘留量的檢測(cè)方法,并進(jìn)行了方法學(xué)驗(yàn)證,現(xiàn)報(bào)告如下。
1 儀器與試藥
Aglient 7890A氣相色譜儀,F(xiàn)ID檢測(cè)器;梅特勒XS205分析天平。三氯甲烷(國(guó)藥集團(tuán)化學(xué)試劑有限公司提供,分析純,批號(hào):20090116),N-N二甲基甲酰胺(DMF)(迪馬公司提供,色譜純,批號(hào):62106)。苦參蒼術(shù)口服液由洛陽(yáng)惠中獸藥有限公司提供,批號(hào):20101001,20101002,20101003。
2 方法與結(jié)果
2.1 色譜條件與系統(tǒng)適用性[3-5]采用DB-FFAP毛細(xì)管柱(30 m ×0.53 mm×1 μm);進(jìn)樣口溫度:220℃;程序升溫,初始溫度40℃保持10 min,以10℃/min的速率升溫至70℃,保持3 min后,再以20℃/min的速率升溫至200℃;FID檢測(cè)器溫度250℃;載氣為氮?dú)猓魉?.0 mL/min;分流比1∶1;進(jìn)樣方式為直接進(jìn)樣;進(jìn)樣量:1 μL。三氯甲烷色譜峰與其他色譜峰的分離度大于1.5,色譜圖見圖1。
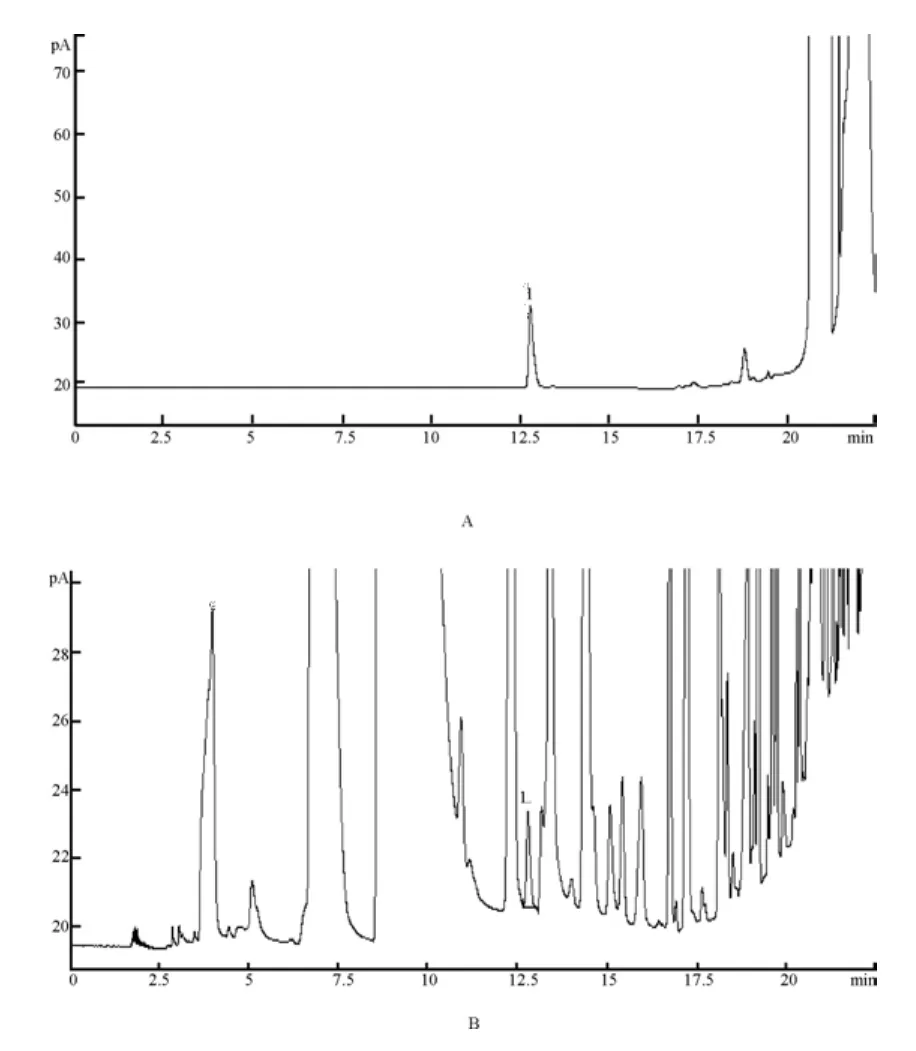
圖1 氣相色譜圖
2.2 對(duì)照品溶液的制備 取三氯甲烷對(duì)照品適量,精密稱定,用DMF稀釋制成24 μg/mL的溶液,作為對(duì)照品溶液。
2.3 供試品溶液的制備 取蒼術(shù)苦參口服液樣品作為供試品溶液。
2.4 線性關(guān)系考察 精密稱取三氯甲烷對(duì)照品151.16 mg,置已有一定量DMF的50 mL量瓶中,加DMF稀釋至刻度,作為對(duì)照品儲(chǔ)備液。分別精密量取對(duì)照品儲(chǔ)備液適量,用DMF稀釋配制成含三氯甲烷6.0464、12.0928、24.1856、30.232、60.464、120.928 μg/mL的系列對(duì)照品溶液,精密吸取上述對(duì)照品溶液各1 μL,注入氣相色譜儀,連續(xù)測(cè)定2次,以平均峰面積(A)為縱坐標(biāo),濃度(C)為橫坐標(biāo)進(jìn)行線性回歸,得回歸方程為 A = 0.9722C-0.1428(r=0.99995),線性范圍為 6.0464~120.928 μg/mL。
2.5 檢測(cè)限和定量限 取濃度為6.046 μg/mL的對(duì)照品溶液,逐步稀釋,依照測(cè)定法測(cè)定三氯甲烷峰的信噪比,得三氯甲烷的檢測(cè)限(S/N=3)為0.91 μg/mL,定量限 (S/N = 10) 為 2.42 μg/mL。
2.6 儀器精密度試驗(yàn) 精密吸取濃度為60.464 μg/mL的三氯甲烷對(duì)照品溶液1 μL,連續(xù)進(jìn)樣6次,測(cè)定其峰面積,計(jì)算RSD為1.1%,表明儀器精密度良好。
2.7 穩(wěn)定性試驗(yàn) 精密吸取供試品溶液1 μL,分別在0,2,6,8,10,12 h 進(jìn)樣,測(cè)定三氯甲烷的峰面積,計(jì)算RSD為2.3%,表明供試品溶液在12 h內(nèi)穩(wěn)定性良好。
2.8 重復(fù)性試驗(yàn) 取批號(hào)為20101001的苦參蒼術(shù)口服液,按上述方法平行測(cè)定6次,得平均含量為22.036 μg/mL,RSD 為2.7%,表明方法的重復(fù)性良好。
2.9 回收率試驗(yàn) 精密量取已知含量的供試品(批號(hào):20101001)5 mL,置10 mL量瓶中,精密加入濃度為22.003 μg/mL的三氯甲烷對(duì)照品溶液5 mL,混勻,共制備6份,按上述方法測(cè)定,結(jié)果見表1。
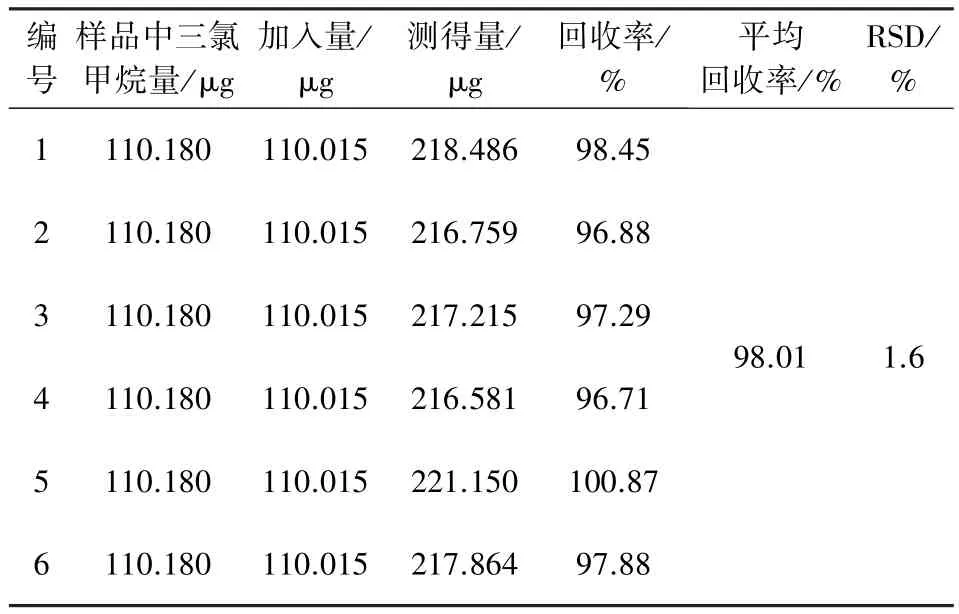
表1 回收率試驗(yàn)數(shù)據(jù)
2.10 供試品的測(cè)定 按“2.1”項(xiàng)下條件進(jìn)行測(cè)定,記錄三氯甲烷峰的峰面積,按外標(biāo)法計(jì)算其含量,結(jié)果3批供試品中均檢出三氯甲烷,其殘留量均低于《中華人民共和國(guó)獸藥典》的限度規(guī)定,數(shù)據(jù)見表2。

表2 樣品的含量測(cè)定結(jié)果 (n=4)
3 討論與小結(jié)
3.1 色譜柱的選擇 研究中分別采用HB-5、DB-624、DB-WAX、DB-FFAP等色譜柱進(jìn)行了色譜條件摸索,結(jié)果表明采用DB-FFAP色譜柱分離效果最好,三氯甲烷峰與相鄰色譜峰的分離度均大于1.5,符合氣相色譜法的規(guī)定。
3.2 檢測(cè)器的選擇 三氯甲烷含有3個(gè)電負(fù)性的氯原子,在FID檢測(cè)器上響應(yīng)較低,最適宜采用電子俘獲檢測(cè)器(ECD)對(duì)其進(jìn)行測(cè)定。ECD檢測(cè)器僅對(duì)那些能俘獲電子的化合物,如鹵代烴、含N、O和S等雜原子的化合物有響應(yīng),但其對(duì)檢測(cè)樣品要求較高。研究中發(fā)現(xiàn),供試品成分較為復(fù)雜且含有一定量的乙醇,極易對(duì)ECD檢測(cè)器造成污染而影響測(cè)定,故最終選用了FID檢測(cè)器。
3.3 進(jìn)樣方式的選擇 研究中分別對(duì)直接進(jìn)樣方式和頂空進(jìn)樣方式進(jìn)行了比較,結(jié)果由于供試品中含有大量的揮發(fā)油和乙醇,采用頂空進(jìn)樣的方式無法達(dá)到減小干擾的目的,反而增加了干擾,故最終采用直接進(jìn)樣的方式。
3.4 測(cè)定方法的選擇 氣相色譜法測(cè)定含量時(shí),內(nèi)標(biāo)法可減少由儀器系統(tǒng)或操作過程引入的誤差,比外標(biāo)法具有更高的準(zhǔn)確度。但由于供試品中含有的揮發(fā)性成分較多,對(duì)測(cè)定的干擾較大,研究中未能從異丙醇、正丁醇、丙酮、正己烷等有機(jī)試劑中選出合適的內(nèi)標(biāo)物,故最終采用外標(biāo)法進(jìn)行測(cè)定。
3.5 柱溫的選擇 試驗(yàn)中分別對(duì)恒溫方式和程序升溫方式進(jìn)行了考察,結(jié)果采用恒溫方式時(shí),三氯甲烷峰不能與其他色譜峰完全分離;采用程序升溫的方式時(shí),三氯甲烷峰達(dá)到基線分離,峰形較好,分析時(shí)間較短;故試驗(yàn)中采用程序升溫的方式進(jìn)行樣品測(cè)定。
本研究首次利用氣象色譜法對(duì)中藥口服液中三氯甲烷的殘留量進(jìn)行了測(cè)定。結(jié)果表明,該方法簡(jiǎn)便、快速、準(zhǔn)確、重現(xiàn)性好,可作為苦參蒼術(shù)口服液中三氯甲烷殘留量的檢測(cè)方法。此外,根據(jù)產(chǎn)品的制劑工藝,本方法亦可用于蒼術(shù)揮發(fā)油和苦參總生物堿中三氯甲烷殘留量的測(cè)定。
[1]鄒 梅,趙生友,王 瑋.氯仿的免疫毒性作用[J].中國(guó)免疫學(xué)雜志,2002,18(3):178-183.
[2]中國(guó)獸藥典委員會(huì).中華人民共和國(guó)獸藥典二○一○年版一部[S].
[3]宋更申,郭 毅,付 焱,等.毛細(xì)管氣相色譜法測(cè)定鹽酸美金剛原料藥中三氯甲烷的殘留量[J].中國(guó)藥房,2010,21(37):3536-3537.
[4]李志剛,李 晶,高 強(qiáng),等.氣相色譜法測(cè)定甲型肝炎滅活疫苗中三氯甲烷殘留量[J].中國(guó)生物制品學(xué)雜志,2010,23(7):778-780.
[5]李雪春,張衛(wèi)國(guó).中草藥制劑中三氯甲烷殘留量的頂空氣相色譜測(cè)定法[J].職業(yè)與健康,2011,27(1):37-38.
