雞肉和雞蛋中金剛烷胺與金剛乙胺殘留檢測UPLC-MS/MS法研究
2014-11-23 03:55:28畢言鋒王亦琳王鶴佳徐士新
中國獸藥雜志 2014年6期
尹 暉,孫 雷,畢言鋒,王亦琳,王鶴佳,徐士新
(中國獸醫藥品監察所,北京100081)
金剛烷胺和金剛乙胺屬于三環胺類抗病毒藥物(化學結構式見圖1),在人類上有廣泛應用。但是,將人用抗病毒藥物用于畜禽,不僅容易導致動物性食品中藥物殘留、動物中毒、動物機體產生免疫抑制和耐藥性等問題,還可導致病毒發生變異,影響動物疫病控制及人用抗病毒藥物的有效性[1-2]。我國農業部2005年第560號公告明確規定金剛烷胺和金剛乙胺等抗病毒藥物在獸醫上禁止使用,美國FDA于2006年也禁止將金剛烷胺和金剛乙胺等人類抗病毒藥物用于畜禽類。因此,急需要建立動物性食品中該類藥物的殘留檢測方法,以加強對該類藥物的殘留監管,有效保護人類健康。目前我國動物性食品中該類藥物殘留檢測方法的報道較少,少數的報道主要集中在液相色譜法[3]、氣相色譜法[4-5]和液相色譜 - 串聯質譜法[6-9]。液相色譜-串聯質譜法具有高效分離和多組分定性、定量的優點,因此成為近年獸藥殘留檢測方法研究的主要方法,本研究以雞肉和雞蛋為試材,建立了同時可以檢測金剛烷胺和金剛乙胺兩種藥物的UPLC-MS/MS檢測方法,以有效解決此類藥物在雞肉和雞蛋中的檢測方法的欠缺,本方法靈敏度高、定性準確、前處理快速簡便,為該類藥物的殘留情況進行有效監管提供了科學依據。
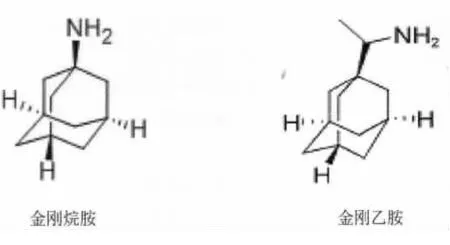
圖1 金剛烷胺和金剛乙胺的化學結構式
1 材料與方法
1.1 儀器 Acquity UPLC-Quattro Premier質譜聯用儀,Waters公司;AE260電子天平,Mettler Toledo公司;Biofuge Strators高速冷凍離心機,賀利氏公司;Organomation Associates氮吹儀,Jnc公司;SIR4漩渦混合器,IKA公司。
1.2 藥品和試劑 金剛烷胺,純度為99%;鹽酸金剛乙胺,純度為99.9%;金剛烷胺 -D6,純度為99.8%,均購自北京振翔科技有限公司;甲酸、乙腈為色譜純,均購自MERCK公司;無水硫酸鈉、正己烷均為分析純;所用水為超純水。
1.3 對照溶液配制 精密稱定金剛烷胺和金剛乙胺對照品適量,置于10 mL棕色量瓶中,用甲醇溶解并稀釋成濃度為1 mg/mL的標準儲備液;量取標準儲備液適量,用甲醇稀釋成10 μg/mL的標準工作液,再用20%乙腈稀釋成100 ng/mL的標準工作液。
1.4 測定方法
1.4.1 色譜條件 色譜柱為BEH C18(50×2.1 mm,1.7 μm),流動相A相為0.1%甲酸乙腈溶液,B相為0.1%甲酸水溶液(梯度洗脫條件見表1),流速0.3 mL/min,柱溫30 ℃,進樣量10 μL。
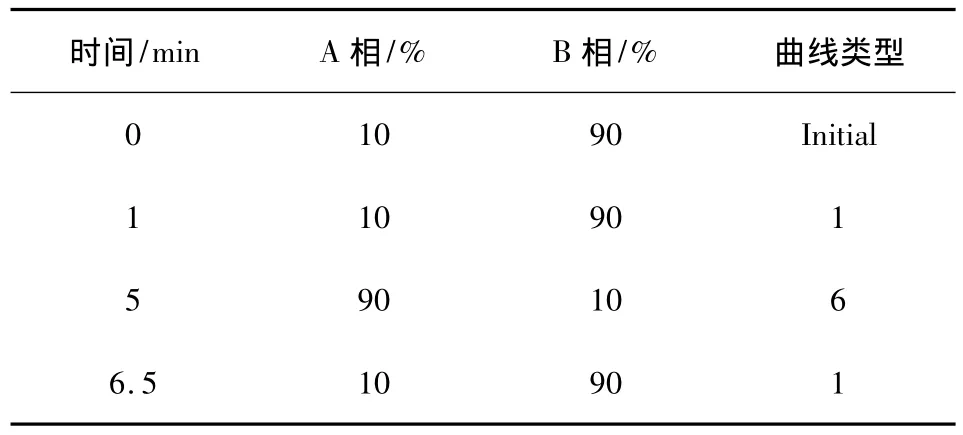
表1 流動相梯度洗脫條件
1.4.2 質譜條件 電噴霧離子源(ESI+),毛細管電壓為3.0 kV,萃取電壓為2.0 V,RF透鏡電壓為0.5 V,源溫為110℃,霧化溫度為350℃,霧化氣速為650 L/h,錐孔氣流速為50 L/h。多反應監測離子情況見表2。
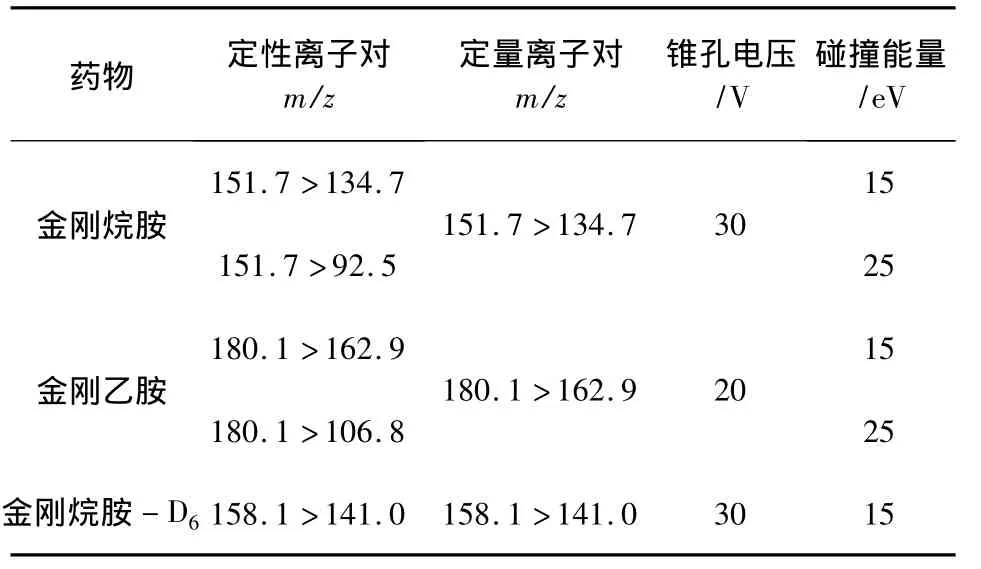
表2 金剛烷胺及內標、金剛乙胺的特征離子、錐孔電壓和碰撞能量
1.4.3 定性與定量 定性時試樣溶液中色譜峰的保留時間,應與校正溶液的保留時間一致,容許偏差為±5%,試樣溶液中的離子豐度比應與校正溶液的一致,容許偏差符合歐盟2002/657/EC決議要求。定量時金剛烷胺采用同位素內標法定量,金剛乙胺通過基質匹配標準溶液采用外標法定量。
1.4.4 標準曲線繪制 分別精密量取適量的金剛烷胺和金剛乙胺標準工作液,制得濃度為2、5、10、20、50、100 ng/mL(含金剛烷胺 -D6為 100 ng/mL)的系列對照溶液,以金剛烷胺特征離子質量色譜峰與內標峰面積比、金剛乙胺特征離子質量色譜峰面積為縱坐標,對照溶液濃度為橫坐標,繪制標準曲線。
1.4.5 樣品前處理過程 稱取(2±0.02)g勻質的雞肉或雞蛋樣品于50 mL離心管內,加3 g無水硫酸鈉,再加入1%甲酸乙腈10.0 mL,渦旋1 min,然后水平震蕩提取10 min,8000 r/min離心8 min,取5.0 mL上清液于溫度50℃下氮氣吹干,加入20%乙腈溶液1.0 mL,充分渦旋溶解,再加入20%乙腈溶液飽和的正己烷2 mL,充分渦旋混合,然后室溫下靜置5 min,吸取下層溶液0.5 mL,轉移至1.5 mL塑料離心管內,12000 r/min離心5 min,取下層清液適量,過0.2 μm濾膜后供UPLC-MS/MS法測定。
1.4.6 方法靈敏度確定 將適量金剛烷胺和金剛乙胺加入到空白雞肉和雞蛋中,制成0.5、1、2 ng/g三個濃度添加樣品,經上述方法進行前處理后,用UPLC-MS/MS檢測,觀察藥物特征離子質量色譜峰信噪比(S/N)和對應藥物濃度,S/N>3者定其為方法的檢測限;S/N>10者定其為方法的定量限。
1.4.7 準確度和精密度的測定 采用標準添加法,在空白雞肉和雞蛋中各添加2、5、10 ng/g三個不同濃度藥物進行回收率試驗,各濃度進行5個樣品平行試驗,重復3次,求批內、批間RSD。
2 結果
2.1 標準曲線 按照上述系列標準溶液濃度進行線性回歸,得到的回歸方程及變異系數見表3。從表中可以看出金剛烷胺和金剛乙胺在2~100 ng/mL濃度范圍內均呈現良好的線性關系,R2分別為0.9977和0.9988。

表3 金剛烷胺和金剛乙胺線性回歸情況
2.2 方法靈敏度 按上述方法進行處理,當添加濃度為2 ng/g時,測得金剛烷胺和金剛乙胺的S/N>10,說明方法定量限為2 ng/g。當添加濃度為1 ng/g時,測得兩種藥物S/N>3,表明方法檢測限為1 ng/g。2 ng/g添加試液中金剛烷胺、金剛乙胺及金剛烷胺-D6特征離子質量色譜圖見圖2、圖3。
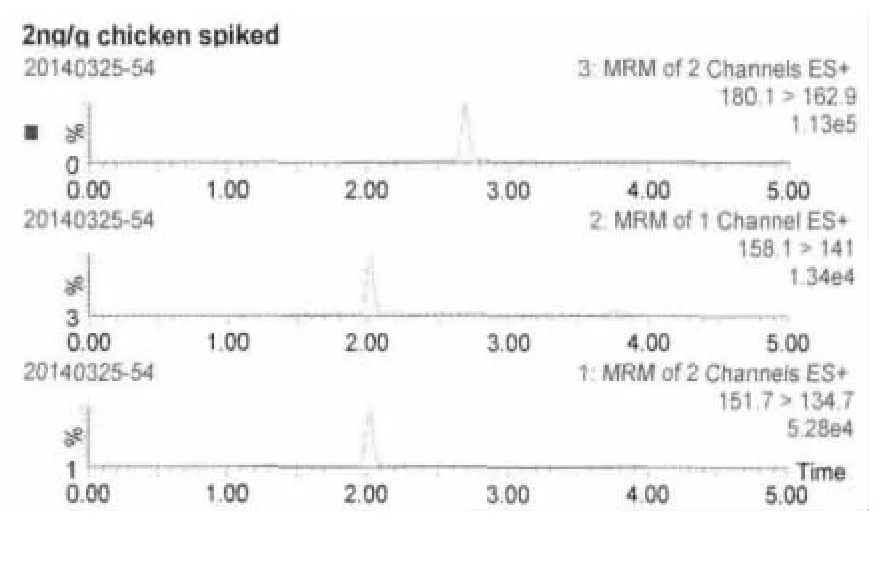
圖2 2 ng/g空白雞肉添加試液中藥物及內標特征離子質量色譜圖
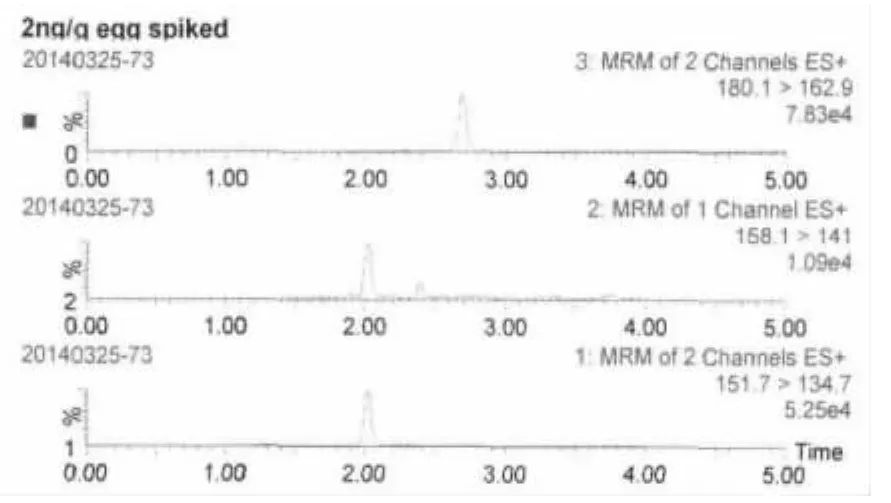
圖3 2 ng/g空白雞蛋添加試液中藥物及內標特征離子質量色譜圖
2.3 方法準確度和精密度 在空白雞肉和雞蛋中各添加三個不同濃度金剛烷胺和金剛乙胺進行回收率試驗,結果匯總見表4、表5。可以看出雞肉中金剛烷胺平均回收率為79.6% ~107.5%,批內RSD為4.4% ~6.8%,批間RSD為6.1% ~7.1%;金剛乙胺平均回收率為78.4% ~101.2%,批內RSD為1.4% ~19.3%,批間RSD為2.5% ~11.3%。雞蛋中金剛烷胺平均回收率為90.8%~111.1%,批內 RSD為3.7% ~11.7%,批間 RSD為7.4% ~9.2%;金剛乙胺平均回收率為75.4% ~87.4%,批內RSD為2.1% ~8.4%,批間RSD為5.0%~6.9%。可以看出,雞肉和雞蛋樣品中兩種藥物的回收率均在70% ~120%之間,批內和批間RSD均小于15%。
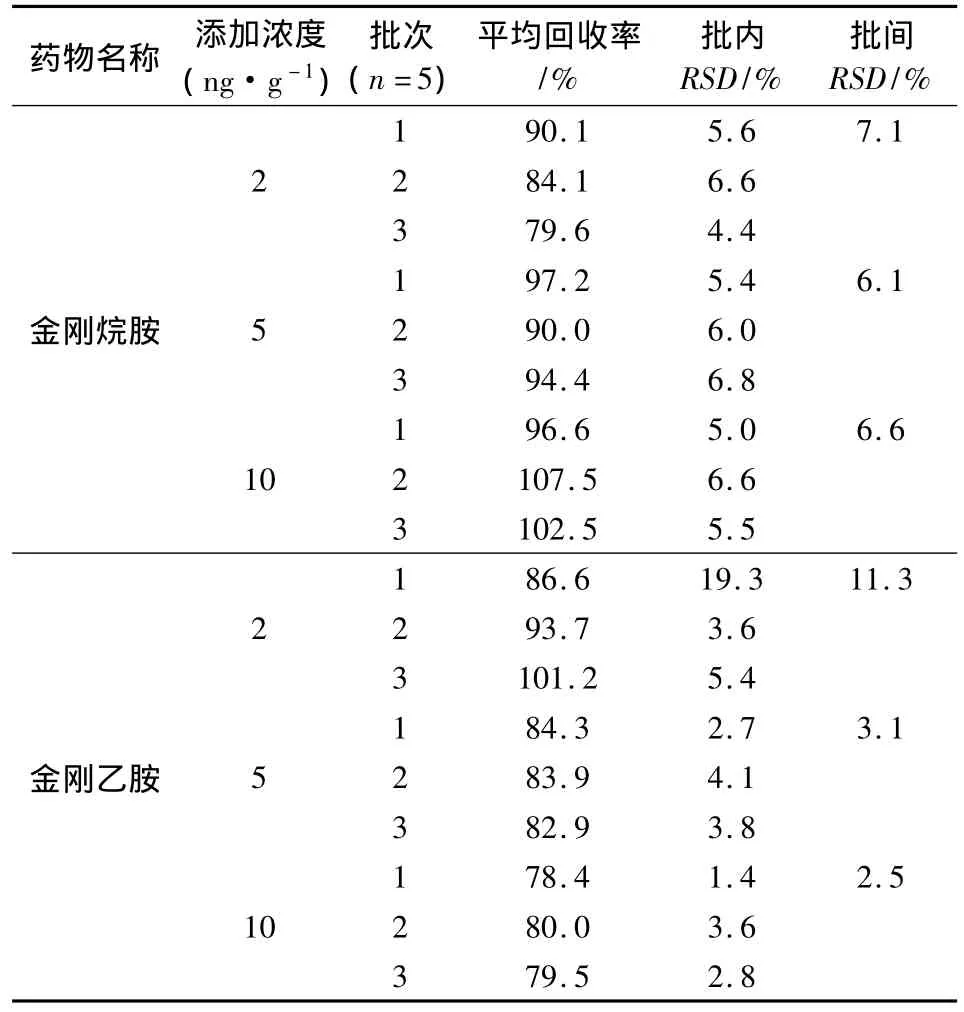
表4 雞肉中金剛烷胺和金剛乙胺添加回收率試驗結果
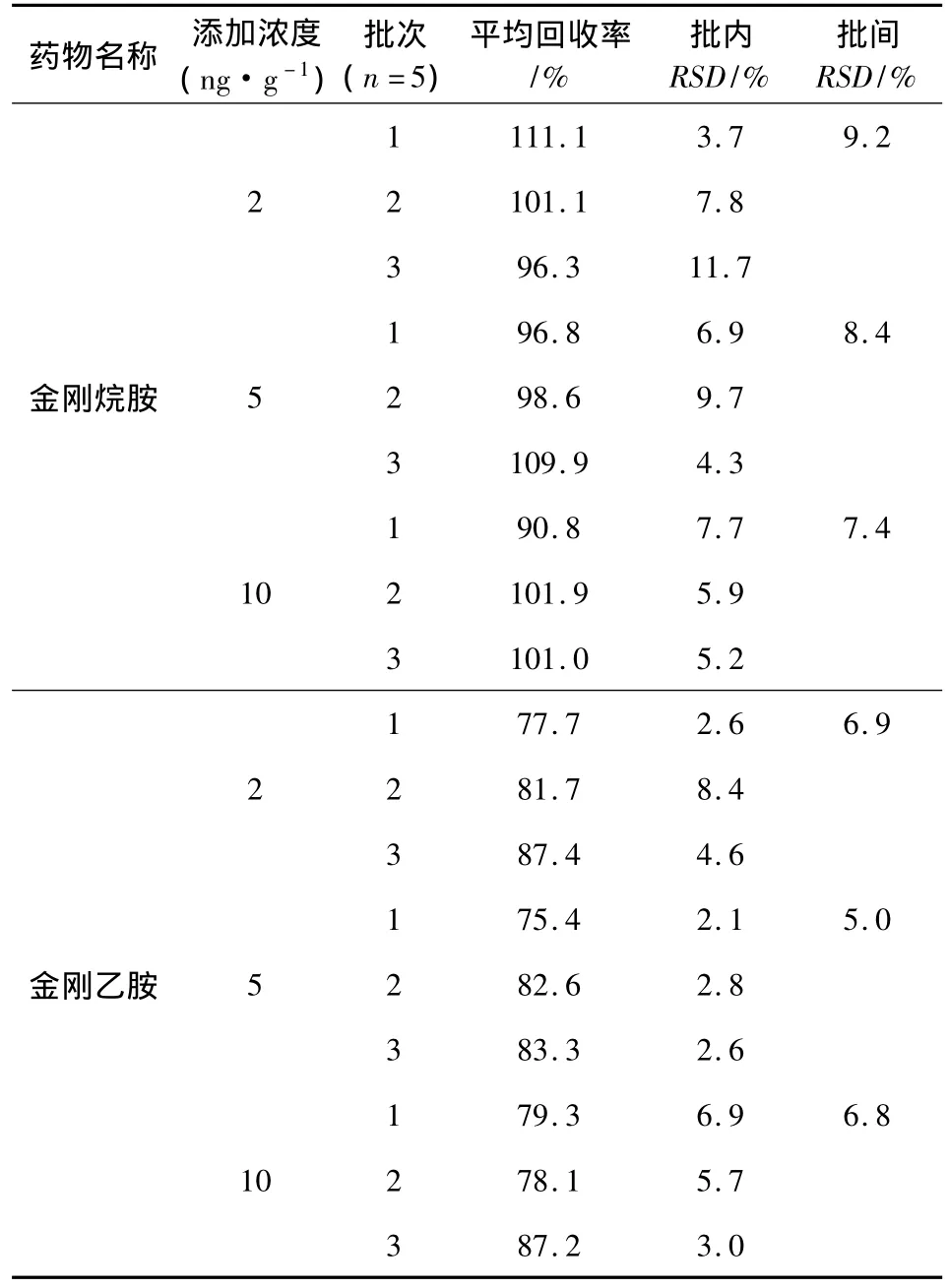
表5 雞蛋中金剛烷胺和金剛乙胺添加回收率試驗結果
3 討論
金剛烷胺和金剛乙胺屬于三環胺類抗病毒藥物,含有氨基,屬于有機堿,在酸性溶液中易溶于水,因此,樣品前處理時采用含有甲酸的乙腈溶液進行提取,用正己烷去除脂肪,采用高速離心的方式進一步去除蛋白質等雜質,通過樣品測試發現凈化效果較為理想,優化了樣品前處理方法。雞肉和雞蛋等動物性食品基質較為復雜,通過充分的提取凈化后該類藥物在質譜檢測時仍存在基質帶來的影響。在流動相中加入0.1%甲酸溶液有利于化合物的離子化,并通過實驗對比確定了最佳洗脫梯度,優化了液相色譜分離條件。通過對方法線性、靈敏度、回收率和精密度等技術參數的考察,發現均能滿足殘留檢測方法的要求,另外,該方法簡便、快速、雜質干擾少,是一種理想的確證檢測方法。
[1]陸學勝,朱天夫,許 敏,等.金剛烷胺對帕金森病患者角膜內皮細胞損害作用的研究[J].世界臨床藥物,2010,31(7):417-421.
[2]葉金朝,葉 菲.美多巴及金剛烷胺誘發精神分裂癥及下肢水腫病案分析[J].藥物流行病學雜志,2002,11(4):219-220.
[3]朱勝平,鐘 華,何 飛,等.高效液相色譜法測定鹽酸美金剛烷胺片的含量[J].中南藥學,2005,3(4):209-210.
[4]徐文彤,劉 紅.氣相色譜法測定硫酸金剛烷胺的含量[J].中國藥師,2007,10(11):1082-1083.
[5]姜大為,范振莉.氣相色譜法測定硫酸金剛烷胺的含量[J].黑龍江科技信息,2012,(7):32-32.
[6]劉正才,楊 方,余孔捷,等.液相色譜-電噴霧串聯質譜法同時檢測雞組織中5種抗病毒類藥物的殘留量[J].色譜,2012,30(12):1253-1259.
[7]李 彥,關麗麗,牟 妍,等.QuEChERS-UPLC-MS/MS檢測畜禽組織中金剛烷胺殘留[J].食品研究與開發,2013,(23):37-40.
[8]陳慧華,韋敏玨,周 煒,等.液相色譜-串聯質譜法測定動物組織中金剛烷胺和金剛乙胺的殘留量[J].質譜學報,2013,34(4):226-232.
[9]魏秀麗,高迎春,陳 玲,等.超高效液相-串聯質譜法測定雞肉組織中金剛烷胺殘留[J].中國獸藥雜志,2013,47(6):53-55.
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
