低溫費-托合成低溫冷凝物加氫精制反應集總動力學
2014-12-31 11:59:50孫啟文劉繼森張宗森吳建民
石油學報(石油加工) 2014年4期
李 志,孫啟文,劉繼森,張宗森,吳建民
(1.上海兗礦能源科技研發(fā)有限公司,上海 201203;2.煤液化及煤化工國家重點實驗室,上海 201203)
近年來,以費-托合成為關鍵技術的煤間接液化工藝備受關注[1]。其中,低溫費-托合成的主產(chǎn)物為冷凝物和石蠟,其具有無硫、無氮、極少芳烴等特點,經(jīng)過加工后可以得到高附加值的汽油、柴油、航空煤油等[2-4]。上海兗礦能源科技研發(fā)有限公司生產(chǎn)的低溫費-托合成冷凝物分為高溫冷凝物(記為L-HC)和低溫冷凝物(記為L-LC)[5]。需要對它們進行深加工,才能獲取汽油、柴油等產(chǎn)品。加氫精制是對費-托合成冷凝物進行加工的重要環(huán)節(jié),目的在于使其中的烯烴飽和,同時脫除含氧化合物,以防止造成后續(xù)加工單元的催化劑失活和反應器腐蝕[6]。目前,關于費-托合成油加氫精制的研究報道不多,并且集中于催化劑開發(fā)和工藝研究方面[7],動力學方面的研究較少。筆者采用Ni-W催化劑在固定床反應器中研究L-LC的加氫精制動力學,旨在為加氫精制反應器的設計以及操作條件的選擇提供依據(jù),同時采用獲得的動力學方程進行分析,以期對反應控制提供依據(jù)。
1 實驗部分
1.1 原料與試劑
L-LC,上海兗礦能源科技研發(fā)有限公司提供;γ-Al2O3、硝酸鎳、偏鎢酸銨,均為分析純,國藥集團化學試劑有限公司產(chǎn)品。
1.2 催化劑的制備
取一定量20~40目的γ-Al2O3置于燒杯中,緩慢滴加已配制好的硝酸鎳和偏鎢酸銨混合溶液,并不斷攪拌。滴加完畢,停止攪拌,浸漬12h。取出固體,干燥,在500℃下焙燒4h,得到Ni-W催化劑。催化劑的BET比表面積163.05m2/g,孔容0.53mL/g,平均孔徑11.05nm。
1.3 實驗流程
采用中國北京欣航盾公司MRT-6212型固定床反應器進行低溫費-托合成低溫冷凝物(L-LC)加氫精制實驗,反應器由φ12mm×1mm的不銹鋼管制成,實驗流程示于圖1。首先,在反應器中裝填2g Ni-W催化劑,通入硫化油(含2%(質(zhì)量分數(shù))CS2的L-LC)和H2進行硫化,H2與硫化油體積比(氫/油體積比)為400。硫化完畢,按動力學實驗要求設定操作條件,通入L-LC和H2進行加氫精制。待反應穩(wěn)定3h,采用配有 HP-PONA 色譜柱的Angilen6890氣相色譜儀分析液相產(chǎn)物的PONA組成。
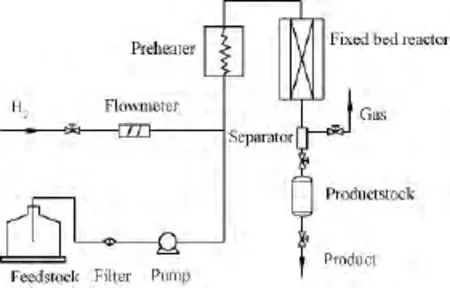
圖1 L-LC加氫精制動力學實驗流程Fig.1 Kinetics experiment flow scheme for L-LC hydrofining
2 低溫費-托合成低溫冷凝物加氫精制動力學方程的建立
2.1 反應網(wǎng)絡及動力學模型的構建
將L-LC加氫精制液體產(chǎn)物劃分為正構烯烴(I1)、異構烯烴(I2)、正構烷烴(I3)、異構烷烴(I4)和含氧化合物(I5)5個集總。wi表示產(chǎn)物中Ii的質(zhì)量分數(shù),定義反應空時(τ)為液時空速(LHSV)的倒數(shù),即τ=1/LHSV。
通過實驗可知,隨著τ的延長,w1和w5不斷降低并趨近于零,w2和w4先增大后減小,中間出現(xiàn)峰值,并且w2比w4更早地出現(xiàn)峰值;w2最終趨近于零,w4趨近于一個較小的值但不是0,w3不斷增加并趨近于一個較大的值但不是100。因此,構建反應網(wǎng)絡時考慮將I2和I4作為中間產(chǎn)物,I3和I4之間存在可逆反應,構建的反應網(wǎng)絡如圖2所示。
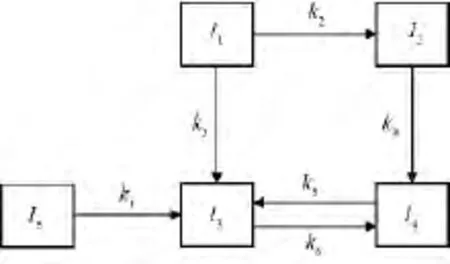
圖2 用于L-LC加氫精制動力學研究的五集總加氫精制反應網(wǎng)絡Fig.2 Five-lumped hydrofining reaction network for L-LC hydrofining
該反應體系中主要為烯烴加氫飽和反應和含氧化合物脫氧反應,因此設定各個反應均為一級反應[8-9],得到動力學方程(1)。
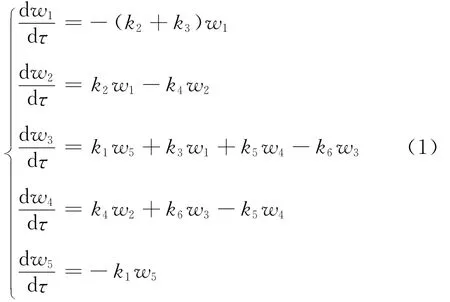
式(1)中,k為圖2中所示的各反應速率常數(shù)。
2.2 動力學模型參數(shù)的計算
在液時空速1~40h-1(即空時0.025~1h)、反應溫度200~300℃、壓力3MPa、氫/油體積比為300的條件下,進行反應動力學實驗,結果如圖3中的離散點所示。在計算動力學模型參數(shù)時,以實驗值與模型計算值之間的殘差作為目標函數(shù),見式(2)。
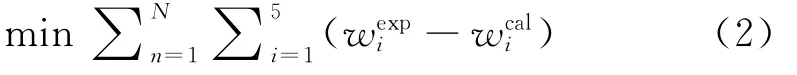
式(2)中,n為實驗序號,N為實驗次數(shù),exp代表實驗值,cal代表模型計算值。
采用Runge-Kutta法對式(2)積分,進而采用Marquardt法計算上述目標函數(shù),得到不同溫度下的反應速率常數(shù),結果列于表1。
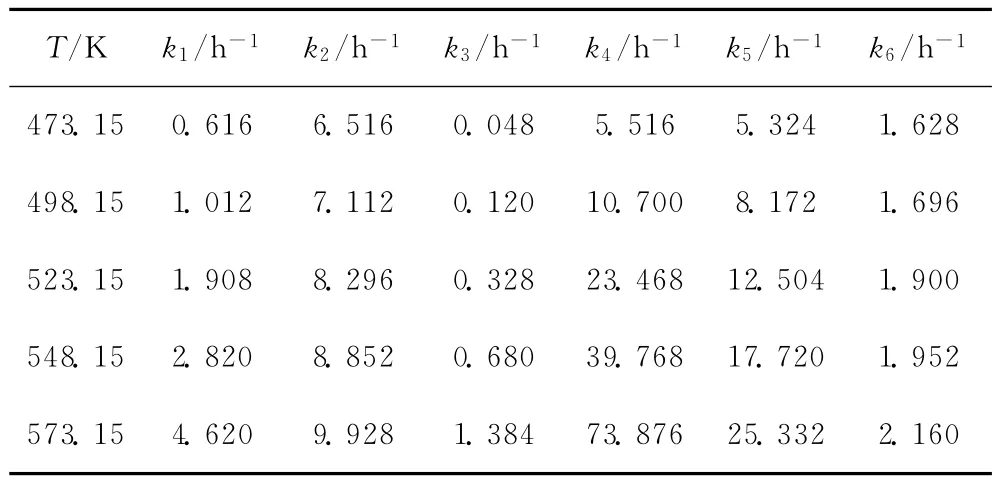
表1 計算所得不同溫度下L-LC加氫精制的反應速率常數(shù)(ki)Table 1 The reaction rate constant(ki)of L-LC hydrofining at different temperatures
速率常數(shù)與反應溫度的關系遵循Arrhenius方程,如式(3)所示。
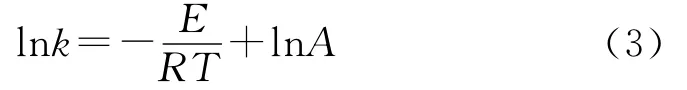
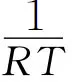
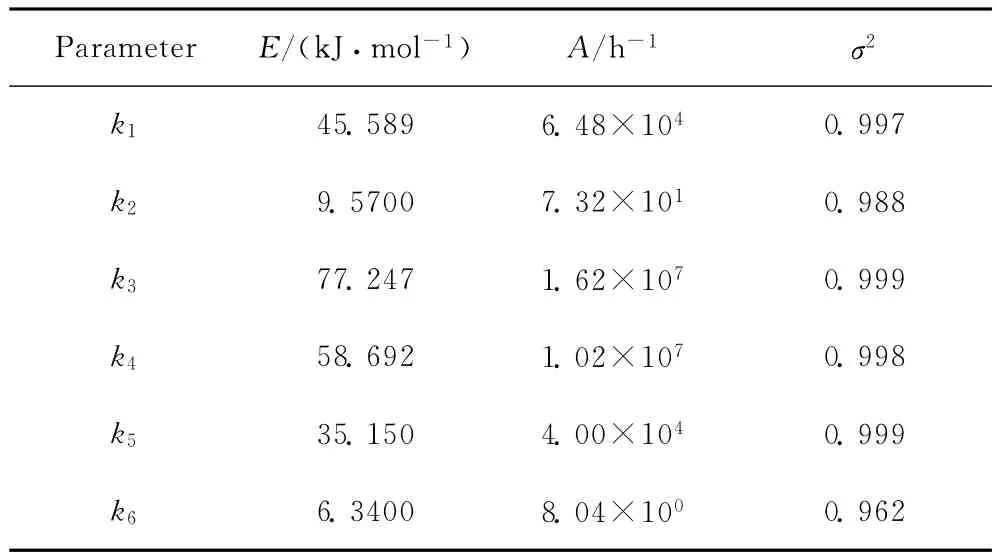
表2 L-LC加氫精制的動力學參數(shù)Table 2 Kinetics parameter values of L-LC hydrofining
將上述動力學參數(shù)代入式(1),可得完整的L-LC加氫精制動力學模型方程(4)。

2.3 動力學模型的檢驗
為檢驗該動力學模型方程的準確性,采用變步長四階 Runge-Kutta法計算式 (4)。初始條件:τ=0,w1=36.89,w2=13.63,w3=32.12,w4=9.81,w5=7.55。模型計算值與實驗值隨空時的變化示于圖3。由圖3可見,實驗值與模型計算值吻合較好,表明該方程是正確的。
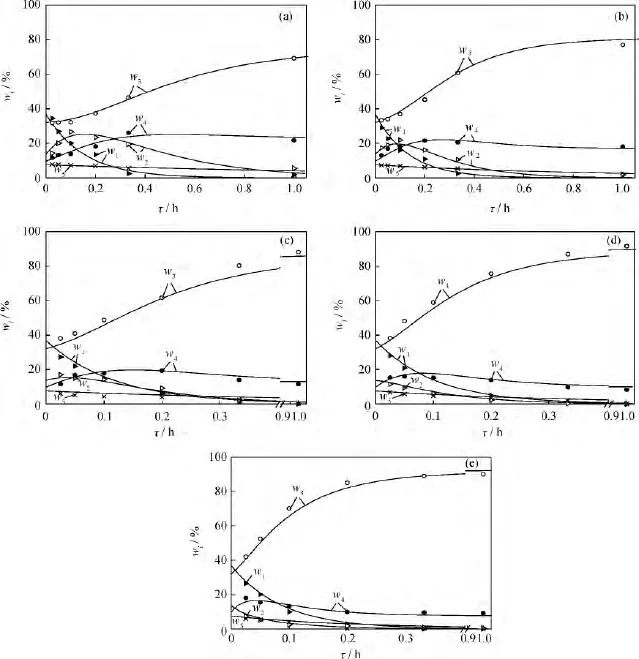
圖3 不同溫度(T)下L-LC加氫精制wi隨空時(τ)變化的實驗值和模型計算值Fig.3 Experimental and calculated values of wivs space time(τ)in L-LC hydrofining at different temperatures(T)
3 結果與討論
3.1 L-LC加氫精制中烷烴異構化反應平衡組成及平衡常數(shù)
由圖2所示的反應網(wǎng)絡可知,當τ→∞時,w2=w5=0,w3和w4分別均趨于一個定值w3,f和w4,f,此時該反應網(wǎng)絡簡化為一個可逆反應(I3I4),稱之為烷烴異構化反應,并且將上述關系代入式(1),可得k6w3,f=k5w4,f,同時w3,f+w4,f=100,因此得到式(5)。
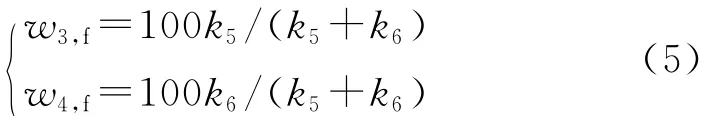
由此可見,L-LC加氫精制產(chǎn)品的平衡組成只與溫度有關。w3,f和w4,f隨溫度變化如圖4所示。定義烷烴異構化反應(I3I4)的平衡常數(shù)K=w4,f/w3,f=k6/k5,其隨溫度的變化也示于圖4。
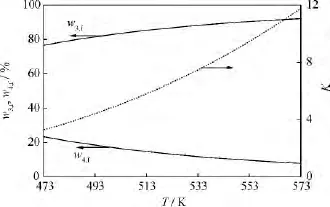
圖4 L-LC加氫精制中烷烴異構化的平衡組成(w3,f和w4,f)及平衡常數(shù)(K)隨溫度(T)的變化Fig.4 w3,f,w4,fand Kof alkane isomerization vs temperature(T)in L-LC hydrofining
3.2 L-LC加氫精制中間產(chǎn)物的動力學規(guī)律
隨著τ的延長,L-LC加氫精制中間產(chǎn)物w2和w4出現(xiàn)峰值,分別記為w2,max、w4,max,其對應的τ分別記為τ1、τ4。由式(4)可以計算得到w2,max、w4,max、τ1和τ4隨溫度的變化,如圖5所示。由圖5可知,隨著溫度的升高,w2,max和w4,max不斷降低,τ1和τ4不斷減小,并且τ1始終小于τ4,即w2比w4更早地出現(xiàn)峰值。I2是I1→I2→I3連串反應的中間產(chǎn)物,并且由于k4活化能幾乎是k2活化能的6倍,I1→I2的反應速率k2比I2→I4的反應速率k4對溫度更加敏感;隨著溫度的升高,I2的生成速率比消耗速率增長得慢,以至于在溫度>543K時,已經(jīng)很難觀察到w2的峰值,因為在溫度較高時I2的消耗速率遠大于I2的生成速率,使得I2剛剛被生成就幾乎被消耗殆盡。然而,因為I4不僅是I2→I4→I3連串反應的中間產(chǎn)物,同時參與I3I4可逆反應,影響w4變化的因素較多。
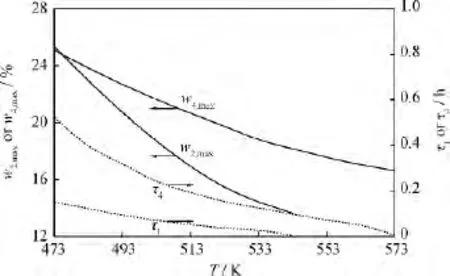
圖5 L-LC加氫精制中間產(chǎn)物w2和w4的峰值及其對應的τ隨溫度(T)的變化Fig.5 The maximum of w2and w4and itsτvs temperature(T)in L-LC hydrofining
3.3 L-LC加氫精制各集總組分的反應速率
L-LC加氫精制各集總組分的反應速率r和反應速率變化率δ可用式(6)表示。
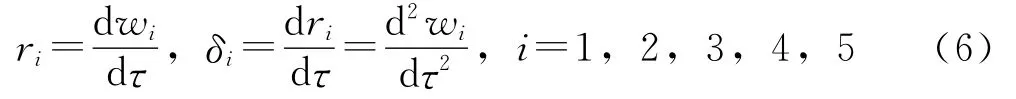
由式(6)可知,當ri>0時,組分Ii總體表現(xiàn)為被生成;當ri<0時,組分Ii總體表現(xiàn)為被消耗。當反應溫度為523.15K,各組分表現(xiàn)的動力學規(guī)律較為典型,因此加以詳細分析,其他反應溫度與之類似。反應溫度為523.15K時,各集總組分反應速率隨空時的變化如圖6所示。由于I5數(shù)值較小對整體反應影響不大,因此圖中未標出。另外,圖中僅標識了4個關鍵空時點。
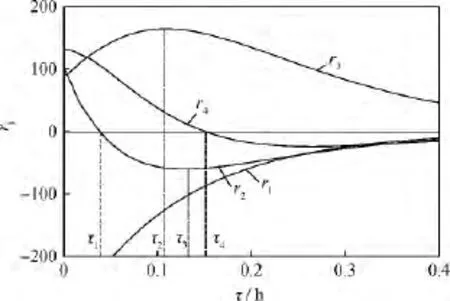
圖6 反應溫度為523.15K時L-LC加氫精制中各集總組分反應速率(ri)隨空時(τ)的變化Fig.6 Reaction rate(ri)variation with space time(τ)at 523.15K
從圖6和式(6)可以看出,(1)對于I1,整個空時區(qū)間內(nèi),r1<0,δ1<0,說明I1一直被消耗,并且I1的消耗速率也不斷降低。(2)I2的反應速率變化最為復雜,當0<τ<τ1時,r2>0,δ2<0,可見I1→I2→I4連串反應中的I1→I2反應相對于I2→I4反應占據(jù)優(yōu)勢,但優(yōu)勢越來越小,這主要是由于開始階段w1較大而w2較小造成的;當τ1<τ<τ3時,r2<0,δ2<0,可見I2→I4反應相對于I1→I2反應占據(jù)優(yōu)勢,并且優(yōu)勢越來越大,這主要是由于經(jīng)歷一段時間的反應后,I1被大量消耗而I2被不斷生成而升高的緣故;當τ3<τ時,r2<0,δ2>0,因為此時w1已經(jīng)很小,所以主要進行I2→I4反應直至w2→0;當τ=τ1(或τ3)時,r2=0(或δ2=0),表現(xiàn)為w2-τ曲線上的1個最高點(或拐點)。(3)對于I3,整個空時區(qū)間內(nèi),r3>0,說明I3始終被生成,當0<τ<τ2時,δ3>0,盡管此時I4不斷增加會推動I3I4反應左移,但是結合表1和圖2可知k3w1?k5w4,可見I3的生成主要源于I1→I3反應;當τ2<τ時,δ3<0,此時由于I1的含量迅速降低促使I4→I3反應成為I3的主要生成反應,而k5w4始終不高,致使r3下降。(4)對于I4,當0<τ<τ4時,r4>0,δ4<0,w2維持在一個較高的水平,促使I2→I4反應比I4→I3反應占據(jù)優(yōu)勢,但優(yōu)勢越來越小;當τ4<τ時,r4<0,δ4逐漸趨于0,I2已經(jīng)較低,w4通過前期的積累會推動I3I4反應左移,直至達到平衡。
4 結 論
采用固定床反應器,進行Ni-W催化劑催化低溫費-托合成低溫冷凝物(L-LC)加氫精制動力學實驗。以實驗結果為依據(jù),將反應產(chǎn)物劃分為5個集總,構建了加氫精制反應網(wǎng)絡,獲得L-LC加氫精制集總動力學模型,該模型的計算結果與實驗結果吻合,驗證了模型的準確性。分析了L-LC加氫精制中烷烴異構化反應的平衡組成,得到其摩爾反應焓為-28.81kJ/mol,并討論了中間產(chǎn)物的動力學規(guī)律和各集總組分的反應速率的變化。
[1]DAVIS B H.Fischer-Tropsch synthesis:Overview of reactor development and future potentialities[J].Topics in Catalysis,2005,32(3-4):143-168
[2]王建平,翁惠新.費-托合成油品的加工利用[J].煉油技術與工程,2006,36(1):39-42.(WANG Jianping,WENG Huixin.Processing and utilization of oil fractions from Fischer-Tropsch process[J].Petroleum Refinery Engineering,2006,36(1):39-42.)
[3]徐倩,左乘基.利用費托合成制取液體燃料的研究進展[J].能源技術,2008,29(4):212-216.(XU Qian,ZUO Chengji.Further research and application of the synthesis of liquid fuel via Fischer-Tropsch synthesis[J].Energy Technology,2008,29(4):212-216.)
[4]CALEMMA V,GAMBARO C,PARKER W O,et al.Middle distillates from hydrocracking of FT waxes:Composition,characteristics and emission properties[J].Catalysis Today,2010,149:40-46
[5]孫啟文.煤炭間接液化[M].北京:化學工業(yè)出版社,2012:505-506.
[6]LAMPRECHT D.Hydrogenation of Fischer-Tropsch synthetic crude[J].Energy & Fuels,2007,21(5):2509-2513.
[7]BOLDER F H A.Fischer-Tropsch wax hydrogenation over a sulfided nickel-molybdenum catalyst[J].Energy&Fuels,2007,21(3):1396-1399.
[8]丁建軍,黃星亮.催化裂化輕汽油中烯烴加氫反應宏觀動力學[J].化學工程,2011,39(9):61-65.(DING Jianjun,HUANG Xingliang.Hydrogenation macrokinetics of olefin in catalytic cracking light gasoline[J].Chemical Engineer(China),2011,39(9):61-65.)
[9]韓崇仁.加氫裂化工藝與工程[M].北京:中國石化出版社,2001:71-75.
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數(shù)理化·中考版(2022年11期)2022-02-16 07:01:20
中學生數(shù)理化·七年級數(shù)學人教版(2020年10期)2020-11-26 08:24:50
數(shù)學物理學報(2020年2期)2020-06-02 11:29:24
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
光學精密工程(2016年6期)2016-11-07 09:07:19
發(fā)明與創(chuàng)新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
核科學與工程(2015年4期)2015-09-26 11:59:03
