實現記憶檢索和回憶中漫游的自傳體記憶模型
2021-09-15 11:20:38劉征
計算機應用與軟件 2021年9期
劉 征
(河南牧業經濟學院信息工程學院 河南 鄭州 450044)
0 引 言
認知模型是人工智能領域研究的重要方向之一,其中自傳體記憶(Autobiographical Memory, AM)是一種編碼、存儲和指導檢索與個人經歷相關的所有事件集信息[1]。自傳體記憶是人類思維的重要組成部分,但國內對自傳體記憶的建模研究較少。回憶通常被認為是自傳體記憶的一種功能,在自我接受和自我改變中起著至關重要的作用。回憶中漫游是指回憶一系列前后相關的自傳體記憶,這些自傳體記憶跨越不同的生活事件,這也是回憶療法的基礎,通常用于改善老年人的心理和認知健康[2]。事件集記憶和自傳體記憶是兩個密切相關的術語,兩者都是指一個人所經歷的過去事件的記憶集合,其中自傳體記憶可以被認為是一種特殊類型的事件集記憶。從個人的角度來看,自傳體記憶包含了一個人一生的經歷,但是現有的大多數計算事件集和自傳體記憶模塊在使用“和/或”表示方式上沒有明顯的區別。
記憶模塊是各種認知模型的重要組成部分。文獻[3]提出的認知模型除了短期“和/或”工作記憶模塊外,還包括長期記憶模塊。這些認知模型可以不指定其所用長期記憶模塊的確切類型,如事件集[4]、語義[5]或自傳體[6]。文獻[7]的特定模型明確地描述了一個事件集記憶模塊的組合,其主要用于通過挖掘存儲的歷史數據來執行基于事例的推理。文獻[8]提出了一種獨立于其他認知模塊的計算事件集記憶模型,并明確定義了計算機游戲中已發生過去事件的形成、檢索和遺忘,但該模型的使用僅限于對歷史數據的回憶,不包含作為輸入字段之一的情緒,而這是自傳體記憶中的一個重要元素。文獻[9]提出的Xapagy自傳體記憶模型被設計為實現敘事推理,其活動大致類似于人類在故事中所表現出來的一些心理過程。Xapagy結合了復雜的自然語言處理方法,但其使用僅限于講故事。文獻[10]開發了一種在線系統,使得用戶能夠基于他們的移動數據,構建出可視化記憶,作為可視化自傳體記憶的一種形式,以便進行自我反省和分享經驗。文獻[11]將人與機器人的交互作用存儲為自傳體記憶,可以使類人機器人積累經驗并提取出規律性。但是在檢索存儲記憶時,前面提到的自傳體記憶模型都僅使用最小數量的索引知識調用簡單的檢索,僅檢索全部記憶、特定用戶的全部記憶,或構成所選動詞的全部記憶。文獻[12]提出了一種基于關鍵詞查詢的自傳體記憶模型用于記憶檢索。該模型以描述游戲環境中發生事件的句子形式輸入記憶,并保存全部已解析關鍵詞的鏈接圖,其中與鏈接相關的權值表示關鍵詞對的共存。而在關于事件集記憶和自傳體記憶模型的文獻中,很少有關于漫游現象的研究。文獻[13]試圖用自陷吸引子神經網絡模仿短期和長期聯想記憶中的漫游效應,并將網絡中的漫游稱為允許“稀疏連接的網絡在遠離初始狀態的吸引子附近徘徊”機制。
不同于前面提到的自傳體記憶模型,本文提出一種可以實現記憶檢索和回憶中漫游的自傳體記憶模型,旨在捕捉記憶,包括一個人生活經歷的圖片快照以及相關的背景,即時間、地點、人物、活動和情緒,可以采用不同類型的線索來檢索編碼的自傳體記憶,并模仿人類思維在回憶中的漫游現象。基于精確的、部分的和含噪記憶檢索線索的實驗結果表明,所給模型具有魯棒的和靈活的記憶檢索,尤其是對于含噪線索的響應具有更好的性能,同時還能通過回憶一系列前后關聯的記憶模仿思維漫游。
1 自傳體記憶模型的心理學基礎
在心理學家建立的各種自傳體記憶模型中,通常從一般到具體將自傳體記憶知識分為3個層次:生命周期、一般事件和特定事件知識,如圖1所示。如果線索是具體和個人相關的,則自傳體記憶可以直接被訪問;如果線索是一般的,則必須生成檢索過程,以得到相關記憶檢索的更多具體線索。直接檢索與生成檢索之間的主要區別是:生成檢索中的控制過程對檢索過程進行調整。這兩種記憶檢索方式之間的差異得到了神經影像學證據的支持。
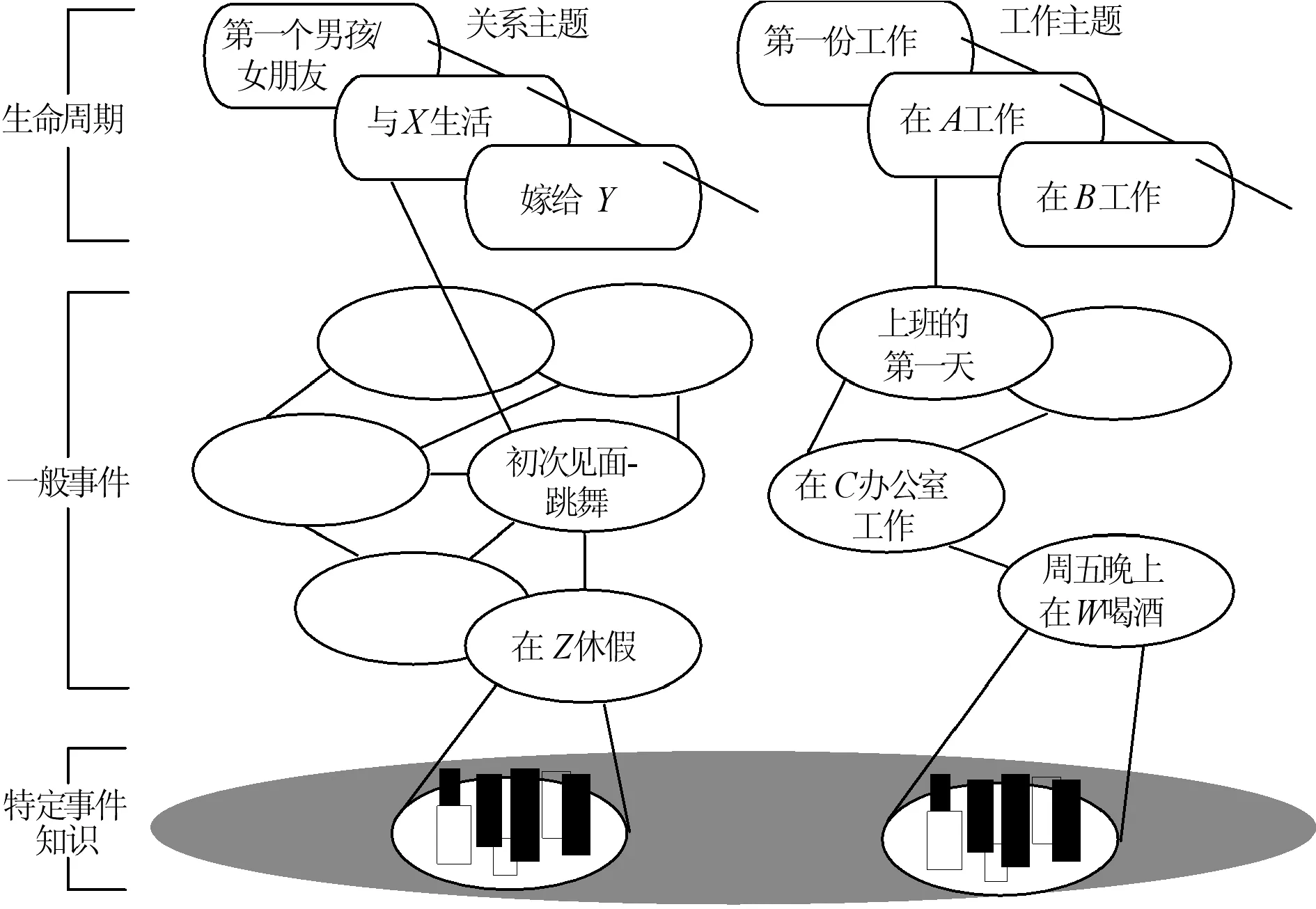
圖1 自傳體記憶層次結構示意圖
2 模型構建及分析
2.1 模型結構
圖2為本文自傳體記憶模型的網絡體系結構。該結構是一個自上而下的3層網絡結構,其中F3層、F2層和F1層分別編碼生命周期、一般事件和特定事件知識3個層次。對比圖1和圖2即可得出它們之間的對應關系,如“在A工作”的生活經歷可表示為F3中的代碼(學習集),與這一事件集的相關事件“上班的第一天”“在C辦公室工作”和“周五晚上在W喝酒”可以表示為F2中的代碼(學習事件),其中一個具體的事件,如“周五晚上在W喝酒”可以在F1中讀出,即周五晚上(時間)、在W(地點)、和同事(人物)、飲酒(活動)、感到快樂(情緒),以及圖像記憶(圖像)。
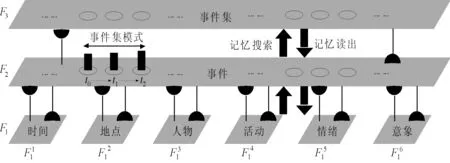
圖2 本文自傳體記憶模型的網絡體系結構
本文模型利用自組織網絡的動態性,其底層將特定事件知識進行編碼,其中特定事件知識由5W1H構成,即時間(何時,When)、地點(何地,Where)、人物(誰,Who)、活動(干什么,What)、意象(哪一種,Which)和情緒(如何,How),中間層通過關聯特定事件知識對事件進行編碼,頂層通過關聯相關事件對事件集進行編碼。自傳體記憶的記憶檢索首先發生在底層,由底層提供檢索線索。按照自下而上的記憶搜索過程,可以分別在中間層和頂層識別相應的事件和事件集,所以可以通過執行自上而下的記憶讀出過程來檢索。
根據圖2所示的F1層(由6個輸入字段構成)和F2層(由1個關聯字段構成),下面給出本文自傳體記憶模型的動態性分析。
關聯字段:令y=(y1,y2, …,yC)表示F2中的激活向量,其中C表示F2中的代碼數。
參數:自傳體記憶的動態性受與下層中全部輸入字段相關聯參數的調節,即選擇參數αk>0、學習速率參數βk∈[0,1]、貢獻參數γk∈[0,1](∑γk=1)和警戒參數ρk∈[0,1]。
代碼激活:自下而上的知識搜索從計算F2中全部代碼的激活(選擇函數值)開始。具體來說,給定xk,對于每個F2代碼j,選擇函數Tj計算如下:
(1)

代碼競爭:代碼競爭過程中,識別具有最高選擇函數值的F2代碼,獲勝者標記為指標J,其中:
TJ=argmax{Tj:對于全部F2代碼j}
(2)
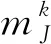
(3)
如果違反任何警戒約束,則會發生失配重置,其中在輸入表示期間將TJ設置為0。因此,另一個F2代碼將被選為新的贏家。這個搜索和評估過程將保證到結束,因為將識別一個滿足警戒標準的已提交代碼或未提交代碼(全部權值被初始化為1以滿足標準)來編碼新的輸入模式。一旦一個未提交代碼被采用,一個新的未提交代碼將自動添加到F2中,然后自傳體記憶模型就可自組織其網絡結構。
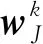
(4)
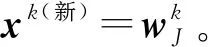
模板掩蔽:由于融合ART的動態性,并不是全部輸入向量都必須提供給知識檢索。在這種情況下,缺失向量xk的所有值(包括補集)都設置為1。
2.2 事件的編碼和檢索
在圖2所給自傳體記憶模型中,F1中的輸入字段分別編碼5W1H。為了使激活向量xk緊湊和通用,采用歸一化值來表示時間和地點,并用明確的值來呈現人物、活動、情緒和意象。

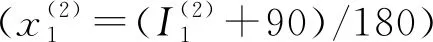
人物向量(x(3)):它是一個二進制值向量,表示參與事件的人。它的長度對應于基于關系的人的分類。
活動向量(x(4)):它是一個二進制值向量,表示事件的內容。它的長度對應于活動的分類。
情緒向量(x(5)):它是一個二進制值向量,表示事件期間的感覺如何。情緒是我們過去經歷中的一個重要組成部分,它影響著自傳體記憶的編碼和檢索。把情緒分為9種,即中性、驚訝、興奮、快樂、滿意、疲倦、悲傷、痛苦和惱怒。因此,x(5)的長度為18。這種分類遵循文獻[15]建立的快樂-喚醒模型。
圖像向量(x(6)):它是一個二進制值向量,表示與事件相關的圖片記憶。它的值被編碼存儲在圖像的文件路徑。在記憶檢索過程中,該向量不與其他向量一起作為檢索線索的一部分。因此,在算法3和算法4中只有F1的前5個輸入字段被調用。
在自傳體記憶模型的F2層編碼事件,事件編碼和檢索過程的偽代碼如算法1所示。
算法1事件編碼和檢索過程的偽代碼
Step1給定輸入模式Ik,在F1中編碼xk;
Step2激活F2中的全部代碼;
//見式(1)
Step3repeat選擇獲勝者代碼J;
//見式(2)
Step4until共鳴發生;
//見式(3)
Step5if需要編碼then執行學習;
//見式(4)
Step6endif
Step8endif
2.3 事件集的編碼和檢索
假設一個事件集的相關事件發生在t0,t1,…,tn,令yti表示發生在ti事件的激活值。則為了編碼事件序列,需要始終保持ytn>ytn-1>…>yt0,因此采用衰減參數τ∈(0,1)來調節激活衰減,使得在每一個新的時間步有yj(新)=yj(舊)(1-τ)。
在自傳體記憶模型的F3層編碼事件集,以關聯F2中編碼的相關事件。其中,事件集編碼和檢索過程的偽代碼如算法2所示。
算法2事件集編碼和檢索過程的偽代碼
Step1for 一個事件集的全部后續事件 do
Step2在F2中選擇關于F1中xk的獲勝者代碼J;
Step3yJ←1;
Step4for 全部F2中事先選擇的代碼 do
Step5yi(新)←yi(舊)(1-τ);
Step6end for
Step7end for
Step8在F3中選擇關于y的獲勝者代碼J′;
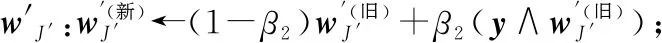
Step10end if
Step12end if
2.4 回憶中漫游
在所給自傳體記憶模型中,漫游是指規則記憶檢索過程,其中后續記憶是基于高度相似但是隨機變化的線索來檢索的。下面討論如何使得所給自傳體記憶模型能夠在回憶中漫游。漫游包括2個主要過程,即在每次迭代中,采用變化的線索,改變檢索線索并迭代地檢索自傳體記憶。
算法3給出改變一個檢索線索過程的偽代碼,在概念上類似于遺傳算法中的染色體變異[16]。所給模型將這一改變過程概括為在給定檢索線索的情況下,有意識地在隨機確定的位置向其添加受調節的噪聲來實現。改變過程受2個參數調節:變異率T∈[0,1]和噪聲級L∈[0,1],采用變異率T來控制某一字段中檢索線索值的變異概率,并用噪聲級L來控制改變量。
算法3記憶檢索線索的變異偽代碼
Step1給定一個記憶檢索線索x={x1,x2,…,x5};
Step2for 全部xi∈xdo
Step3ifrand()≤Tthen
//選第i個字段作為變異

Step5ifi≤2&&rand()≤2/|xi| then
//對于歸一化向量,選擇第j個值進行變異

Step8else ifi≤3&&rand()≤2(1+L)/|xi| then
//對于二值向量
Step10end if
Step11end for
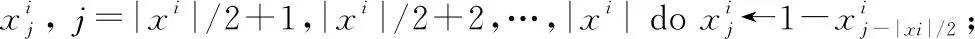
//計算補集
Step13end for
Step14end if
Step15end for
算法4給出所給自傳體記憶模型在回憶中漫游過程實現的偽代碼。其中漫游過程為:在每次迭代開始時給定的記憶檢索線索基礎上,首先使用給定的線索來檢索最相關的事件,該事件尚未包含在檢索的記憶集中,然后將檢索到的事件追加到記憶集中,再用變化的檢索線索進行下一次迭代。算法4的終止準則是通過檢索一個預先確定的事件數量N。這個準則可以很容易地刪除或修改,以便進行連續檢索。
算法4自傳體記憶模型在回憶中漫游過程實現的偽代碼
Step1給定一個記憶檢索線索x={x1,x2,…,x5};
Step2ρk←0,k=1,2,…,6;
//在記憶檢索過程中移除全部警戒標準
Step3M=?;
Step4repeat
Step5repeat 在F2中識別關于x的E;
//尋找獲勝者事件
Step6yE←0;
//抑制其激活值
Step7untilE?M
Step8M←M∪{E};
//保存在M中的檢索順序
Step9變異x;
//見算法3
Step10until |M|=N;
Step11檢索M中的全部事件
3 實 驗
實驗數據主要由我國某體育明星53個事件的快照以及相應的背景構成。53個事件是由12個事件集構成,每個事件集包含3至7個事件。從在線網頁直接提取了除情緒外的所有特征,情緒是從圖片及其背景中手工提取出來的。根據所收集的數據集,定義8種類型人物關系:家庭、鄰居、配偶、朋友、同學、同事、熟人和陌生人,以及15類活動:餐飲、休閑、旅游、度假、購物、夜間外出、娛樂、體育、鍛煉、工作、聚會、社交、慶祝、婚禮和學校。在對輸入向量進行形式化處理后,將數據樣本提交至本文自傳體記憶模型中,在F2中對53個事件進行編碼(采用算法1),在F3中對12個事件集進行編碼(采用算法2)。實驗采用的自傳體記憶參數如表1所示,其中大多數都是采用標準參數值,在實驗中不需要調整。
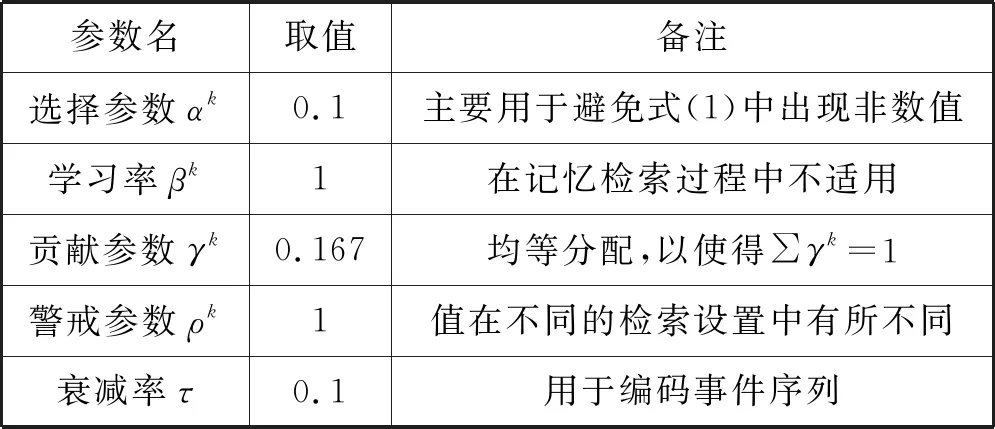
表1 實驗采用的自傳體記憶模型參數
3.1 采用精確、部分和含噪線索的記憶檢索
采用本文自傳體記憶模型對記憶進行編碼后,首先采用以下3種類型的線索測試記憶檢索的性能:
(1) 精確線索:從數據集中隨機選取一個事件,并采用其表示向量x=(x(1),x(2),…,x(5))作為精確檢索線索;
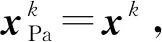
(3) 含噪線索:在所選取的xPa字段中(其中xPa≠(1,1,1,1,1)),給定一個部分線索xPa和含噪線索級L′,引入噪聲來得到含噪線索xNo。將噪聲引入檢索線索的過程遵循算法3中描述的從Step 4到Step 13的過程,其中L=L′。
選擇基于關鍵字的查詢方法作為比較基準,該方法被許多現有照片或記憶存儲庫所采用。其檢索準則是基于給定的檢索線索是否與存儲的記錄的相應部分完全匹配。實驗中在執行基于關鍵字的查詢對給定的檢索線索做出響應后,將對一組事件進行檢索。如果最初用于生成給定線索的事件可以在檢索集中找到,則認為檢索是成功的,否則,認為是失敗的。
本文自傳體記憶模型處理含噪線索時,可以降低警戒參數ρk,這樣用戶即使提供了不完整的線索,也可以檢索某些記憶,這個特性是目前許多其他現有照片或記憶存儲庫類型應用所不具備的優勢。為了響應給定的檢索線索,本文自傳體記憶模型檢索一組記憶并將它們回放給用戶。對于每個實驗,重復10次,每次隨機選擇或生成檢索線索,并將結果匯總,以供進一步分析。
由于采用生成部分線索和計算成功檢索的準則,因此本文自傳體記憶模型和基于關鍵字的查詢方法對精確和部分檢索線索的響應都獲得了100%的成功率;但處理含噪線索時卻很有挑戰性,因為不確定性雖然可以通過我們的大腦很好地進行處理,但不一定能通過許多計算模型來處理。圖3為本文自傳體記憶模型和基于關鍵字查詢方法對不同線索完整性百分數P和不同含噪線索級L′的含噪線索的記憶檢索響應實驗結果。
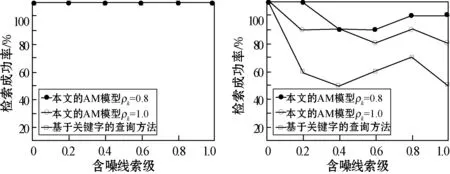
(a) P=0 (b) P=20
當P=0時,所給方法都能檢索數據集中的全部事件,因此在圖3(a)中都達到100%的成功率。但隨著P值的增大,檢索線索由越來越多的輸入字段構成,因此,隨著線索完整性百分數P值和含噪線索級L′值的增大,本文自傳體記憶模型和基于關鍵字查詢方法的記憶檢索性能都普遍下降。同時可以看出,本文自傳體記憶模型明顯在對含噪線索響應的成功檢索率方面表現得更好。通過降低在處理含噪線索時的警戒值,本文自傳體記憶模型可以更好地處理檢索過程中出現的不確定性。因此,在處理不完整信息方面,本文模型更傾向于追求類似于人的智慧,表現出更好的性能。
3.2 在回憶中漫游
為了測試本文自傳體記憶模型在回憶中漫游的性能,采用表2給出的變異率T和噪聲級L的不同組合用于不同設置中作為在回憶中漫游性能的比較。選取T=0.2或T=0.4,可以在變異過程中使期望線索的1個或2個字段分別按平均值改變,選取L=0.1或L=0.2在漫游過程中生成比較小的噪聲。同時,還測試了極端情況WSS和WRR,即T和L設置為邊界值。
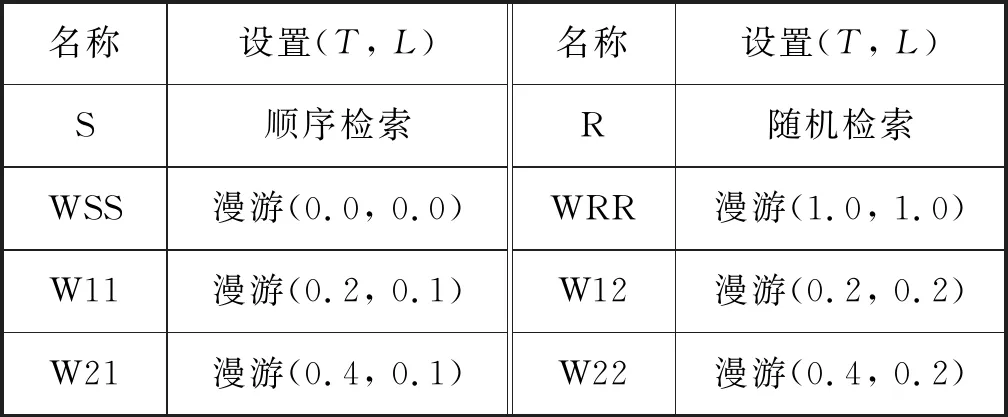
表2 實驗參數設置的命名約定
表3為本文自傳體記憶模型在W22配置漫游過程檢索到的部分記憶序列,其圖像回放如圖4所示。表3很好地說明了本文自傳體記憶模型從第5集(包括5個事件)到第10集(包括7個事件)的漫游,然后在2步后漫游返回第5集。
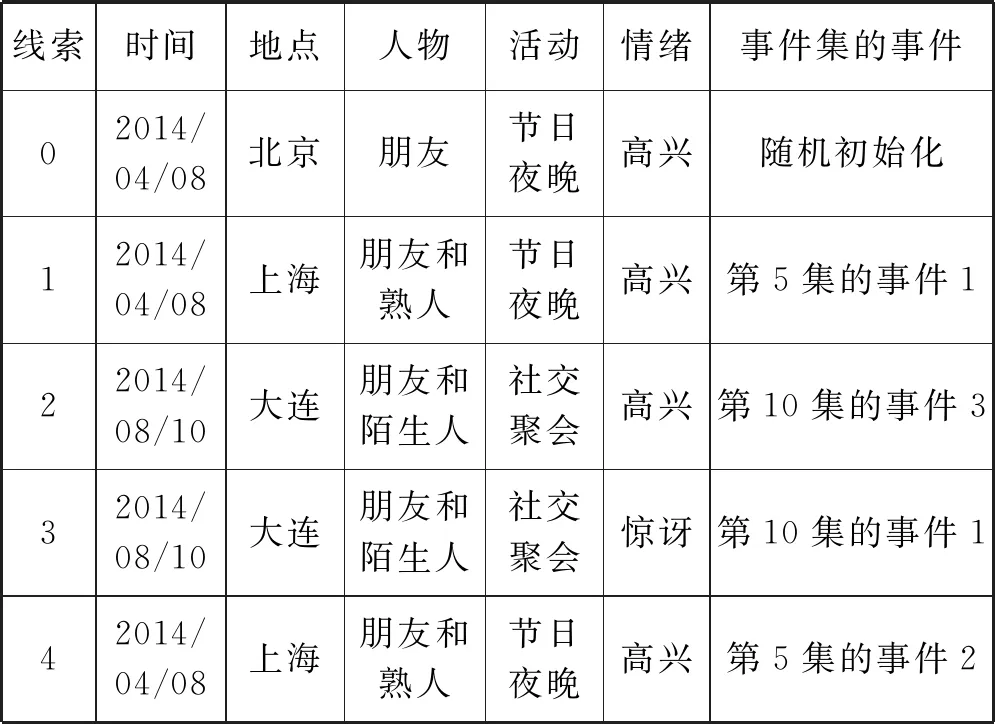
表3 檢索到的自傳體記憶部分子序列
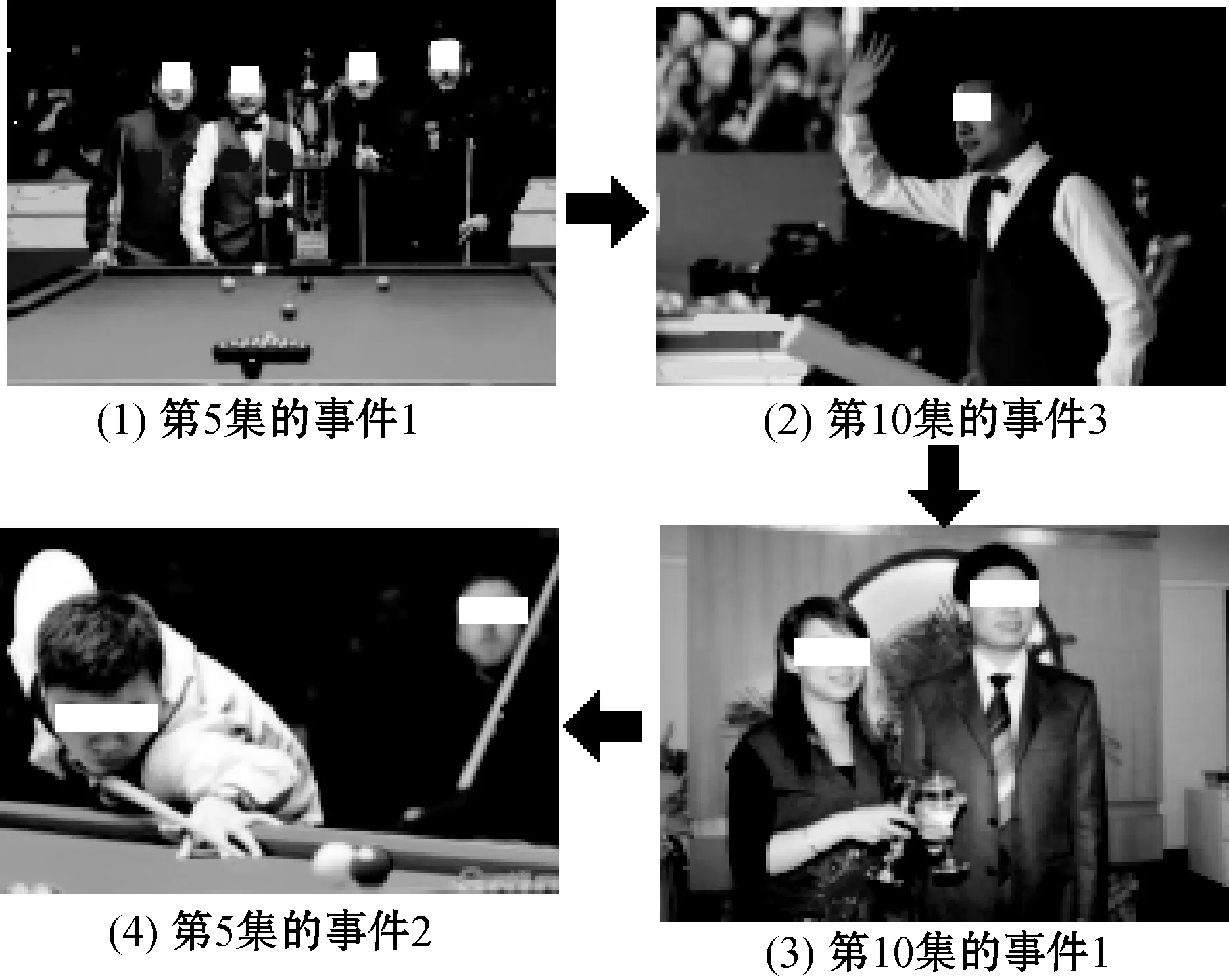
圖4 根據表3所示順序檢索的事件圖像回放
由此可知,本文自傳體記憶模型能夠有效地檢索一個人的自傳體記憶的一個適度子集,并且能夠模仿在回憶中漫游的現象。
4 結 語
為了捕捉自傳體記憶,本文給出一種可實現記憶檢索和回憶中漫游的自傳體記憶模型。該模型不僅用于建模用戶生活經歷的在線自主主體中自傳體記憶的編碼和檢索,而且還能夠模仿人類自傳體記憶中的心靈漫游現象。由實驗結果表明,本文自傳體記憶模型在對含噪線索的記憶檢索方面的性能優于傳統基于關鍵字的查詢方法,這是因為后者無法處理許多現有照片或記憶存儲庫中的含噪線索。同時本文自傳體記憶模型能夠實現在回憶中漫游,可以模仿一個人跨越不同事件集的前后相關聯的記憶序列,即一個人自傳體記憶的一個適度子集。下一步將對本文自傳體記憶模型的感知、識別和推理能力進行重點研究,以期能夠提升本文自傳體記憶模型的實際應用價值。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
學苑創造·A版(2020年10期)2020-11-06 05:21:26
數學物理學報(2020年2期)2020-06-02 11:29:24
作文周刊·小學一年級版(2016年27期)2017-06-03 23:21:17
光學精密工程(2016年6期)2016-11-07 09:07:19
絲綢之路(2016年9期)2016-05-14 14:36:33
新湘評論·下半月(2016年4期)2016-05-05 22:12:41
新湘評論·下半月(2016年4期)2016-05-05 22:12:41
海外文摘(2016年4期)2016-04-15 22:28:55
