HPLC-PDA法測定17種獸藥中非法添加多西環素
2015-03-10 10:11:15周紅霞李慧素吳寧鵬高瑞霖
中國獸藥雜志 2015年10期
關鍵詞:檢測
周紅霞,李慧素*,吳寧鵬,高瑞霖
(1.河南省獸藥飼料監察所,鄭州 450008;2. 鄭州市第七中學,鄭州 450000)
?
HPLC-PDA法測定17種獸藥中非法添加多西環素
周紅霞1,李慧素1*,吳寧鵬1,高瑞霖2
(1.河南省獸藥飼料監察所,鄭州 450008;2. 鄭州市第七中學,鄭州 450000)
建立了17種獸藥中非法添加多西環素的HPLC-PDA法。采用十八烷基鍵合硅膠為填充劑,以醋酸鹽緩沖液-乙腈(80∶20)為流動相,二極管陣列檢測器進行全波長(200~400 nm)掃描,檢測波長為280 nm,并通過液相色譜保留時間、紫外光譜信息和峰純度檢查,對非法添加物質進行定性鑒別和定量測定。結果表明,該色譜條件下,多西環素與其他物質峰分離良好。多西環素在5~500 μg/mL濃度范圍內線性良好,回收率在86.5%~118.1%之間,RSD為0.2%~4.5%,檢測限為0.5mg/g。本方法準確、可靠、重現性好,可用于獸藥制劑中多西環素的定性和定量檢測。
多西環素;峰純度檢查;高效液相色譜法
多西環素(Doxycycline,DOC)又名強力霉素,屬于第二代半合成四環素衍生物,臨床上廣泛用于治療畜禽等動物的細菌性疾病[1]。由于多西環素價格相對低廉、來源廣泛,且抗炎療效明顯,因此極易被不法分子摻入在抗菌消炎類藥物增強療效。通過對市場上銷售和生產的抗菌藥物調研,結合2014年監督抽檢篩查結果,發現市場上存在為提高藥物療效而在部分抗菌藥物中非法添加多西環素的現象,嚴重影響了獸藥市場的穩定。由于非法添加通常是隱形行為,容易造成藥量疊加,帶來耐藥性和藥物殘留的安全隱患,并能通過食物鏈傳遞,對人造成危害[2]。
為了更好的打擊非法添加行為,需要建立獸藥中非法添加多西環素的檢查方法,目前,還沒有對獸藥中非法四環素類中的一種或者幾種藥物進行測定的方法,因此,本試驗參考相關文獻[3~5],采用高效液相色譜法與二極管陣列檢測器,建立了氟喹諾酮類制劑、氟苯尼考注射液及硫酸慶大霉素注射液等17種制劑中非法添加多西環素的HPLC-PDA檢測方法。
1 材料
1.1 儀器與試劑 Agilent 1100高效液相色譜系統,包括二極管陣列檢測器;MILIPORE超純水器;METTLER XP205電子天平。超聲波清洗器:KQ-3200E型。甲醇為色譜純,其他均為分析純,水符合實驗室一級水要求。
1.2 藥品 多西環素對照品,含量為100.0%,批號H0160704,來自中國獸醫藥品監察所。鹽酸多西環素,含量為90%,由河南某獸藥生產企業提供。
甲磺酸達氟沙星注射液、氧氟沙星注射液、甲磺酸培氟沙星注射液、恩諾沙星注射液(溶液)、鹽酸環丙沙星注射液、 煙酸諾氟沙星注射液、氟苯尼考注射液(溶液)、硫酸慶大霉素注射液、甲磺酸培氟沙星可溶性粉(顆粒)、乳酸諾氟沙星可溶性粉、恩諾沙星可溶性粉、氧氟沙星可溶性粉、鹽酸環丙沙星可溶性粉和氟苯尼考粉,均來自市售產品,經檢測均不含多西環素。
2 方法與結果
2.1 色譜條件 色譜柱:Ageal Venusil XBP C18(4.6 mm×250 mm,5 μm);流動相:醋酸鹽緩沖液-乙腈(80∶20),其中醋酸鹽緩沖液(0.25 mol/L醋酸銨- 0.1 mol/L 乙二胺四醋酸二鈉-三乙胺(100: 10 : 1),用冰醋酸或氨水調節p H 值至8.8;二極管陣列(PDA)檢測器,采集波長范圍為200~400 nm,分辨率為1.2 nm;記錄280 nm波長處的色譜圖;柱溫30 ℃;進樣量20 μL。在該色譜條件下,多西環素主峰與各物質峰分離度均符合要求。
2.2 溶液配制
2.2.2 供試品溶液 稱取(量取)供試品1.0 g(或1.0 mL),置50 mL容量瓶中,加0.01 mol/L鹽酸溶液10 mL(氟苯尼考類制劑需加10 mL乙腈),超聲處理5 min,靜置,用0.01 mol/L鹽酸溶液稀釋至刻度,搖勻。精密量取5 mL,置50 mL量瓶中,用0.01 mol/L鹽酸溶液稀釋至刻度,搖勻,作為供試品溶液。
2.2.3 陰性空白溶液配制 稱取(量取)無非法添加的各制劑約1.0 g(或1.0 mL),照2.2.2項下操作制備成陰性空白溶液。
2.2.4 空白添加溶液 取無非法添加的各制劑,按1%、2%、5%進行添加,照2.2.2項下操作制備成各制劑的空白添加溶液。
2.3 峰純度檢查 對供試品溶液色譜圖中多西環素色譜峰進行峰純度檢查,采用被測物峰純度角小于純度閾值的方法確定該被測物為單一物質峰。
2.4 方法學考察
2.4.1 線性 精密量取2.2.1項下對照品貯備液,用0.01 mol/L鹽酸溶液分別稀釋成5、10、20、50、100、200、500 μg/mL的系列標準工作溶液,按照2.1項下色譜條件,從低濃度到高濃度依次注入高效液相色譜儀測定。以對照品溶液濃度為橫坐標、峰面積為縱坐標繪制標準曲線,求得線性回歸方程及相關系數。
2.4.2 回收率試驗 精密稱取多西環素對照品10、50、100 mg各3份,分別置50 mL容量瓶中,加入1.0 g(或1.0 mL)陰性樣品,混勻,加0.01 mol/L鹽酸溶液10 mL溶解(氟苯尼考類制劑需加10 mL乙腈),超聲處理5 min,靜置,用0.01 mol/L鹽酸溶液稀釋至刻度,搖勻。精密量取5 mL,置50 mL量瓶中,用0.01 mol/L鹽酸溶液稀釋至刻度,搖勻,得到相當于多西環素20、100、200 μg /mL添加濃度的溶液,精密量取20 μL,注入高效液相色譜儀,按照2.1項下液相條件記錄色譜圖。計算回收率及及相對標準偏差。
2.4.3 檢測限 將多西環素對照品溶液進行梯度稀釋后,制成系列濃度0.1、0.2、0.5、1、2、5 μg/mL,添加到陰性空白制劑中,照2.2.2項下操作并進行檢測。以光譜圖失真的最大濃度作為方法的檢測限。
例3 (2018年桂林中考第11題)如圖5,在正方形ABCD中,AB=3,點M在CD的邊上,且DM=1,△AEM與△ADM關于AM所在的直線對稱,將△ADM按順時針方向繞點A旋轉90°得到△ABF,連接EF,則線段EF的長為________.
2.4.4 專屬性 通過陰性空白試驗排除各制劑中主要成份對于被測物的干擾。采用被測物峰純度角小于純度閾值的方法進行峰純度檢查。結果顯示,此液相條件下多西環素出峰處無其他干擾峰,為單一物質峰,方法可行。
2.4.5 耐用性 從柱溫、流動相pH值、流速、色譜柱三個方面考察方法的耐用性。改變柱溫分別為25℃、30℃和35℃),調節流動相pH值分別為8.6、8.8和9.0),改變流動相比例分別為75:25、80:20和82:18;改變流速分別為0.8、0.9、1.0 mL/min;選擇不同品牌色譜柱,Agilent ZORBAX SB-C18(4.6×250 mm,5 μm);Thermos Hypersil Gold (4.6×250 mm,5 μm);Waters SunfireTMC18(4.6×250 mm,5 μm),對其峰型,分離度,峰純度和出峰時間進行考察。
2.5 結果與分析
2.5.1 峰純度檢查 對供試品溶液色譜圖中多西環素峰進行峰純度檢查,結果顯示,硫酸慶大霉素注射液和氟苯尼考類制劑均無紫外吸收,對多西環素檢出無干擾,氟喹諾酮類制劑雖有紫外吸收,但與添加物多西環素均能很好的分離,且多西環素峰的純度角度均小于純度閾值,表明在此液相條件下,多西環素的出峰處無其他干擾峰,為單一物質峰,方法可行。

圖1 供試品(氟苯尼考注射液)添加多西環素的色譜圖
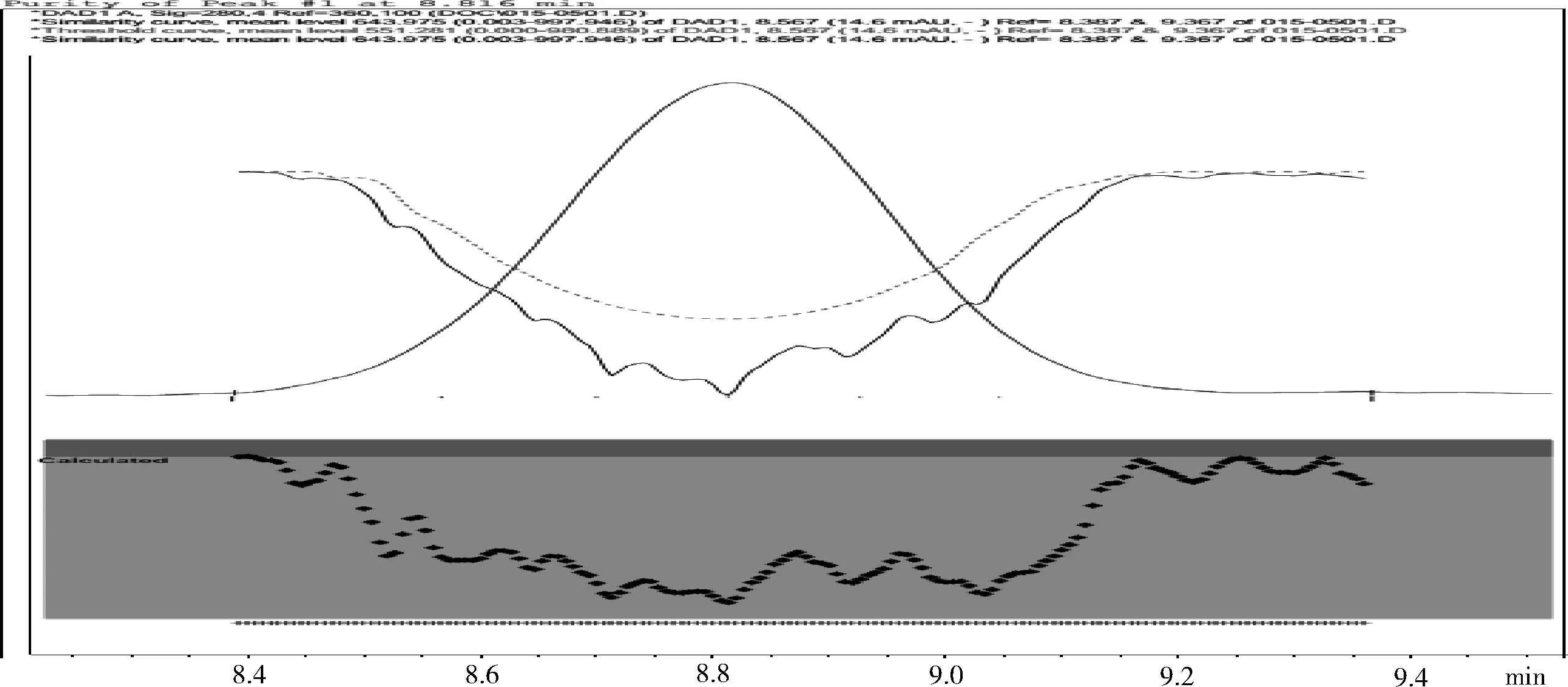
圖2 供試品(氟苯尼考注射液)添加多西環素的純度檢查圖

圖3 供試品(諾氟沙星可溶性粉)添加多西環素的色譜圖
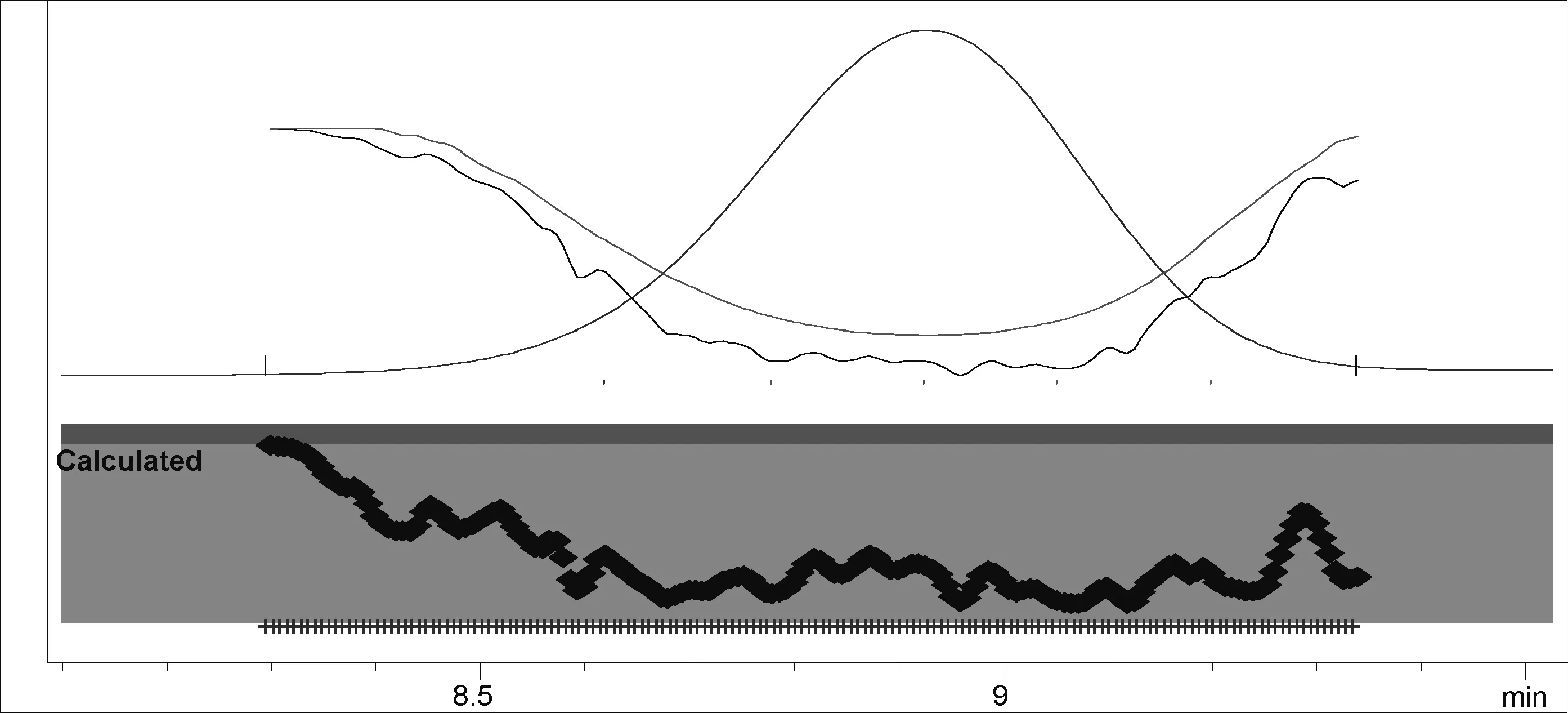
圖4 供試品(諾氟沙星可溶性粉)添加多西環素的純度檢查圖
2.5.2 線性 多西環素在5~500 μg/mL的系列濃度內進行測定,以峰面積(y)對濃度(x)進行線性回歸,多西環素的回歸方程Y=14.801X+44.897和相關系數r為0.9997。結果表明:甲氧芐啶在5~500 μg /mL的濃度范圍內線性關系良好。
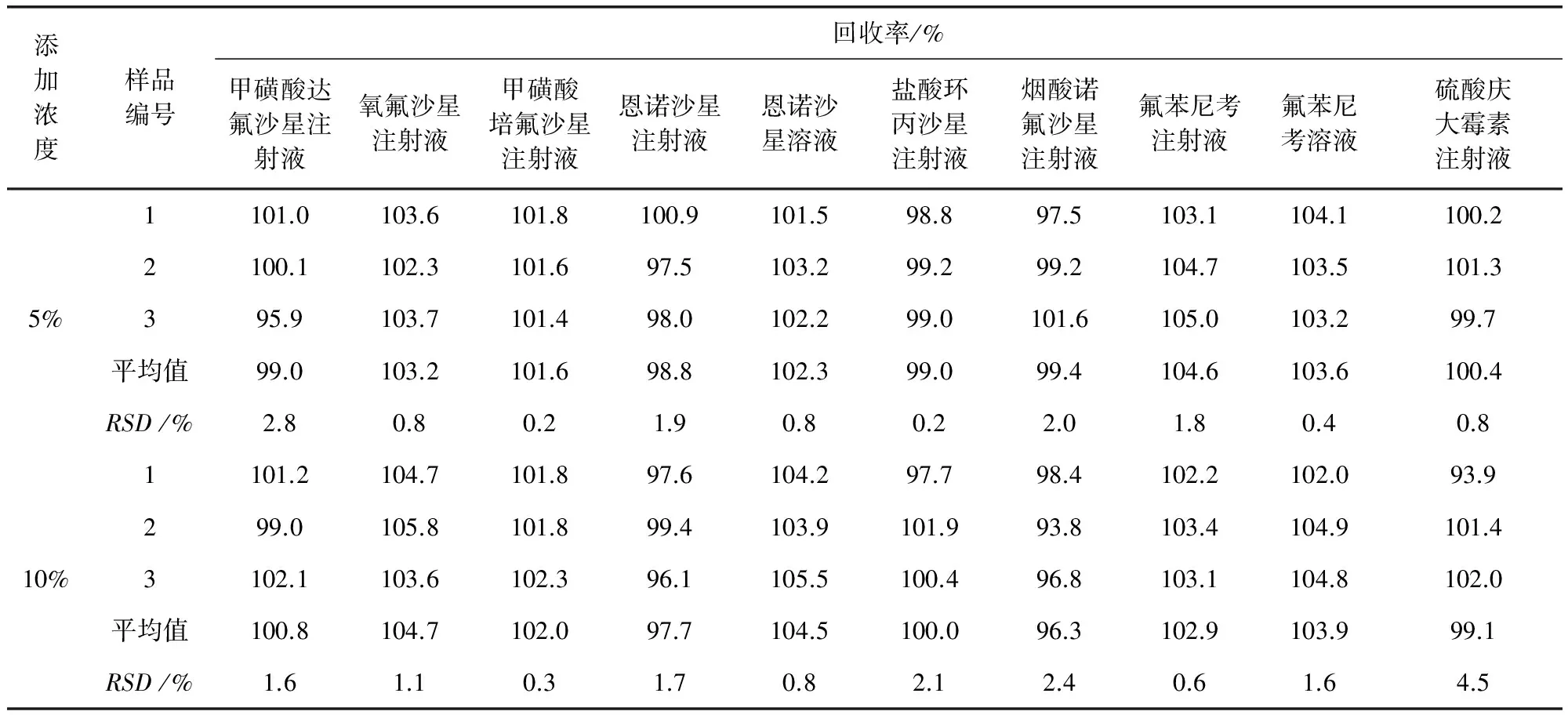
2.5.3 回收率試驗 由于添加回收試驗中多西環素對照品的使用量較大,用符合純度要求的多西環素原料藥代替對照品進行添加回收試驗,即在陰性空白樣中分別添加多西環素原料,按照1%,2%和5%三個濃度進行添加,通過試驗計算回收率及相對標準偏差。該方法中各制劑中添加甲氧芐啶的回收率為86.5%~118.1%,RSD為0.2%~4.5%,結果見表1、表2。

表1 注射液及溶液中添加多西環素回收率試驗結果表
續表

表2 可溶性粉及顆粒添加回收率實驗結果表
2.5.4 檢測限 綜合考慮色譜圖峰型、峰面積和多西環素有效濃度等因素,以光譜圖失真(最大吸收波長差異大于±2 nm)的最大濃度作為方法的檢測限。結果表明,濃度為0.5 μg/mL, 光譜圖失真;濃度為1 μg/mL,光譜圖不失真。
2.5.5 耐用性 通過對柱溫、流動相比例、流速和色譜柱的進行改變,多西環素保留時間受柱溫影響較小,隨柱溫升高,保留時間略提前;隨流動相有機相比例的增加,多西環素的保留時間前移,但對分離度并未產生明顯的影響;隨著流速的增加,保留時間明顯提前;三種不同品牌色譜柱均可用于該檢查,僅保留時間不同。結果顯示,本方法耐用性較好,能夠滿足檢測要求。
3 討論
3.1 檢測波長的選擇 利用二極管陣列檢測器,在190~400 nm波長范圍內進行紫外吸收圖譜掃描,多西環素在280 nm波長處有最大吸收,對所選的17種獸藥制劑進行紫外掃描發現,在280 nm附近都不會產生干擾,所以選擇280 nm為檢測波長。
3.2 流動相選擇 參考相關文獻[6-8],分別比較了0.1 mol/L磷酸二氫銨:乙腈:甲醇 (58:20:22),0.05 mol/L草酸銨溶液-N,N二甲基甲酰胺-0.2 mol/L磷酸氫二銨溶液(65:30:5)和醋酸鹽緩沖液(0.25 mol/L醋酸銨-0.1 mol/L 乙二胺四醋酸二鈉- 三乙胺(100:10:1)-乙腈(80:20),發現采用醋酸鹽緩沖液-乙腈(80:20)時,多西環素峰型對稱,與各供試品主成分分離情況良好。因此選擇了醋酸鹽緩沖液-乙腈(80:20)為流動相。
3.3 檢測限 本實驗采用的HPLC-DAD法測定獸藥制劑中非法添加多西環素,以光譜圖失真的最大濃度作為本方法的檢測限,根據現有儀器條件,當多西環素的上機濃度低于1 μg/mL時,光譜圖已嚴重失真。則多西環素在各制劑中的檢測限為0.5 mg/g(或mg/mL)。
3.4 回收試驗的影響因素 由于本試驗采用的是空白添加的方法,非法添加的稱樣量將直接影響回收率的結果。本實驗添加濃度為1%時,非法添加的稱樣量較少(10 mg左右),采用稱量紙直接稱重法,稱量誤差較大,造成回收率偏低或者偏高。另外,由于多西環素屬于酸堿兩性物質,在酸性溶液(pH<2)、中性或堿性(pH>7)均易發生降解,某些制劑的輔料和主藥與多西環素互相作用也可能會影響回收率的結果,這還待進一步考察。
3.5 稀釋溶劑的選擇 多西環素在水或甲醇中易溶,但多西環素的水溶液極不穩定,在弱酸性溶液中相對穩定,因此,選擇0.01 mol/L鹽酸溶液作為稀釋劑。同時,由于氟苯尼考類在水中微溶解,用0.01 mol/L鹽酸溶液溶解時,會出現白色沉淀,造成非法添加成分溶解不完全;加入10 mL乙腈,可以使取樣量的氟苯尼考類制劑完全溶解,再加入0.01 mol/L鹽酸溶液稀釋時,不會有沉淀析出。
4 結論
本文建立了HPLC-DAD法檢測17種獸藥制劑中非法添加多西環素的檢測方法,結合液相色譜保留時間和紫外光譜兩方面的信息,結合峰純度檢查,增強了檢測結果的準確度,實現獸藥中非法添加物的高效、快速、準確識別。該方法具有操作簡便、回收率高、靈敏度高等特點,可以用于獸藥制劑中多西環素的檢測,并為打擊獸藥制劑非法添加多西環素制假行為,提供了有力的技術支撐。
[1] 中國獸藥藥典委員會.獸藥使用指南(化學藥品卷)[M].北京:中國農業出版社,2010.
[2] 周媛,貢玉清,邵德佳.有關中獸藥中非法添加化學藥物的探討[J],中獸醫醫藥雜志,2010,4(3):28-30.
[3] 丁祥軍. 多西環素分析方法研究進展[J]. 數理醫藥學雜志, 2008,(2):115-116.
[4] 劉福艷, 李曉燕,黃金麗,等. HPLC-DAD法同時檢測抗炎類中藥制劑中非法摻入的6種四環素類抗生素[J]. 中國抗生素雜志,2013,38(7):531-535.
[5] 董玲玲,范強,楊星,等. 氟苯尼考粉中非法添加煙酰胺和氨茶堿的HPLC-PDA檢測方法的建立[J].中國獸藥雜志, 2014,48(5): 47-50.
[6] 李永梅. HPLC法對氟尼康中鹽酸多西環素及氟苯尼考含量的測定 [J]. 中國當代醫藥, 2009,16(13):68-70.
[7] 李向超,黃顯會,曾振靈.多西環素注射液的研制及其含量測定 [J]. 中國畜牧獸醫, 2011,38(8):217-220.
[8] 劉智鈞,張玉蓮. 鹽酸多西環素含量測定方法的比較 [J].廣東化工, 2012,39(7): 188-189.
(編輯:侯向輝)
Determination of Doxycycline in Veterinary Drug by HPLC-PDA
ZHOU Hong-xia1, LI Hui-su1*, WU Ning-peng1, GAO Rui-lin2
(1.HenanInstituteofVeterinaryDrug&FeedControl,Zhengzhou450008,China; 2.ZhengzhouNO.7MiddleSchool,Zhengzhou450000,China)
A method for the determination of doxycycline in 17 kinds of veterinary drug was developed by the high performance liquid chromatography with photo-diode array detector (HPLC-PDA). It was tested with C18 column, using acetate buffer solution - acetonitrile (80∶20) as the mobile phase. The detection wavelength was set at 280 nm and simultaneously collected the spectrum at wavelength of 200~400 nm. After separated by HPLC. The ultraviolet spectra inforrnation, retention time and peak purity test were used to identify and determinate the doxycycline by comparison with those of reference substances.Results showed that doxycycline and other materials finely separated. And Doxycycline showed good linearity in the concentration range of detection. The average recoveries of the drugs ranged from 86.5%~118.1% at spiked levels withRSDless than 4.5%. the detection limit of doxycycline was 0.5 mg/g. In conclusion, the method is accurate and reproducible, and provides a reliable way for detecting doxycycline adulterated in veterinary drug.
doxycycline; peak purity test; HPLC-PDA
2015年國家畜禽產品質量安全風險評估項目(GJFP2015008)
周紅霞,研究員,從事獸藥質量監測與監督。
李慧素。E-mail:lihuisu2010@126.com
2015-08-07
A
1002-1280 (2015) 10-0036-06
S859.83
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
