Rh的芳基化合物與含氮芳基鹵代物交叉偶聯(lián)反應(yīng)機理研究
2015-03-22 10:27:58王曉嵐徐銘瑤胡慶仟李來才
原子與分子物理學報 2015年6期
王曉嵐, 徐銘瑤, 胡慶仟, 李來才
(四川師范大學化學與材料學院, 成都 610066)
Rh的芳基化合物與含氮芳基鹵代物交叉偶聯(lián)反應(yīng)機理研究
王曉嵐, 徐銘瑤, 胡慶仟, 李來才
(四川師范大學化學與材料學院, 成都 610066)
利用密度泛函理論(DFT)研究了Rh的芳基化合物與含氮芳基鹵代物交叉偶聯(lián)過程催化循環(huán)的微觀反應(yīng)機理.在B3LYP/6-31+G(d) 基組水平上(Rh、I采用了贗勢基組LanL2DZ)優(yōu)化了反應(yīng)過程中所有化合物的幾何構(gòu)型并計算了頻率,通過能量、頻率和振動方式確定了中間體和過渡態(tài)的真實性.此外,在同等基組水平上還運用了分子中的原子理論討論了成鍵臨界點的電荷密度的變化,運用了自然鍵軌道理論討論了鍵的性質(zhì)與軌道間的相互作用.為了提高計算精度, 在6-311++G(d,p) 基組水平上計算了反應(yīng)機理中所有物質(zhì)在氣相及溶劑化下的單點能, 得到與 6-31+G(d) 基組計算相同的結(jié)論.結(jié)論表明Rh(Ⅰ)起到了有效的催化作用,且計算所得結(jié)論與實驗結(jié)果相符合.
密度泛函理論; 芳香化合物; 含氮芳基鹵代物; Rh(Ⅰ)催化; 交叉偶聯(lián)反應(yīng); 微觀反應(yīng)機理
1 引 言
C—C鍵形成的偶聯(lián)反應(yīng)在有機合成領(lǐng)域中是重要的研究內(nèi)容之一[1-3].高效高選擇性的C—C偶聯(lián)反應(yīng)的新反應(yīng)和新途徑能夠大大提高目標產(chǎn)物的合成效率,對有機合成化學的應(yīng)用和發(fā)展具有很大的應(yīng)用價值和理論意義.構(gòu)造C—C鍵成鍵的偶聯(lián)反應(yīng)效率由于有機合成化學中的周環(huán)反應(yīng)[4]和過渡金屬催化的偶聯(lián)反應(yīng)的發(fā)展而得到極大提高[5,6].鹵素原子因為可以改變分子的空間結(jié)構(gòu)和親水親油性,從而影響整個化合物分子的物理屬性、化學性質(zhì)和生物活性而在有機合成中占有一個重要的位置.同時鹵代芳烴化合物中的鹵素官能團能通過親核加成、親核取代、氧化加成等反應(yīng)轉(zhuǎn)化為其他官能團,進而改變化合物的化學性質(zhì),達到預期合成效果.因此作為化學原料以及醫(yī)藥中間體中重要的一份子,芳基鹵代物被廣泛使用[7-10].由Rh的配合物來作催化劑的有機鹵代物和金屬有機化合物的交叉偶聯(lián)反應(yīng)的反應(yīng)機理的研究比較少見[11-14].Rh的配合物作為催化劑在有機合成反應(yīng)具有可選擇性高,活性很好,所需反應(yīng)條件比較溫和等特點,已經(jīng)廣泛運用于精細工業(yè)、石油工業(yè)、醫(yī)藥化工等領(lǐng)域[15-21].但作為一種稀有金屬,Rh的回收循環(huán)再利用很重要.Ryo Shintani等已經(jīng)在實驗室里運用Rh配合物作為催化劑完成了多組份有機物的偶聯(lián)反應(yīng)[22].因此本文想通過研究Rh(Ⅰ)催化下的Rh芳基化合物與含氮芳基鹵代物偶聯(lián)反應(yīng)的催化循環(huán)的理論研究,分析其最可能實現(xiàn)的反應(yīng)路徑,更清晰得了解其微觀反應(yīng)機理,為類似的后續(xù)實驗起到一定的指導作用.
2 計算方法
本文對Rh(Ⅰ)催化下的Rh芳基化合物與含氮芳基鹵代物偶聯(lián)反應(yīng)的催化循環(huán)過程中所有的反應(yīng)物、中間體、過渡態(tài)和產(chǎn)物利用密度泛函理論的B3LYP方法,在6-31+G (d) 基組水平上(Rh和I采用了贗勢基組LanL2DZ)進行了結(jié)構(gòu)優(yōu)化,并在相同基組水平下對各構(gòu)型進行了頻率計算.反應(yīng)公式如下:

運用自然鍵軌道(NBO)方法分析了所有物質(zhì)的軌道間相互作用[23].使用自洽反應(yīng)場(SCRF)極化連續(xù)模型(IEF-PCM)模擬實驗所使用的二氧六環(huán)(dioxane)溶劑效應(yīng), 采用6-311++G (d, p) 基組水平對反應(yīng)機理中各化合物均進行了全參數(shù)優(yōu)化,并使用相同基組計算了氣相條件下的單點能.為了進一步的了解成鍵性質(zhì)分析成鍵特征,采用AIM2000的程序包計算了對應(yīng)的所有成鍵臨界點(BCP)和成環(huán)臨界點(RCP) 電荷密度,顯示分子中原子的屬性[24].所有計算均采用Gaussian09程序完成[25].
3 反應(yīng)機理和能量分析
實驗報道中, 在反應(yīng)條件篩選時使用的溶劑為二氧六環(huán)(dioxane), 因此在進行反應(yīng)機理的理論研究時選用與實驗一致的dioxane作為溶劑.對在6-31+G(d) 基組下優(yōu)化后的反應(yīng)物、中間體、過渡態(tài)以及生成物的構(gòu)型使用大基組6-311++G(d,p) 進行了單點能計算,氣相和溶劑化條件下通過計算所得能量(E)、相對能量(Erel)及過渡態(tài)頻率數(shù)值均已列于表1,能量(E)已包括零點能(ZPE)修正.
Scheme 1、2即為此偶聯(lián)反應(yīng)可能存在的計算模擬過程圖,過程中還存在一個含炔烴的芳烴偶氮的芳基化交叉偶聯(lián)反應(yīng)過程.將本文計算模擬出的可能的反應(yīng)通道scheme1、2與文獻相比較[22],可以看出本文與Ryo Shintani所推斷的反應(yīng)過程相一致.整個偶聯(lián)反應(yīng)過程中所涉及的所有反應(yīng)、中間體、過渡態(tài)及產(chǎn)物的構(gòu)型全部列于圖1及圖3中.
3.1 反應(yīng)通道一
在推斷的反應(yīng)通道一中,具體流程圖見Scheme1.Rh插入鹵素之間發(fā)生氧化加成反應(yīng),發(fā)生金屬轉(zhuǎn)移,促使芳基上的C與炔基上的C發(fā)生交叉偶聯(lián),金屬繼續(xù)發(fā)生轉(zhuǎn)移,最后通過還原消除反應(yīng),催化劑順利脫落.
反應(yīng)物Ra與Rh的化合物Ca發(fā)生反應(yīng)后,Ca中的Rh原子插入反應(yīng)物的苯環(huán)和I原子之間生成中間體IM1,體系的總能量降低了295.11 KJ/mol.IM1構(gòu)型是穩(wěn)定存在,催化劑的插入過程較易完成.
IM1經(jīng)由過渡態(tài)TS1生成IM2,這是一個分子內(nèi)的插入反應(yīng)過程. 反應(yīng)活化能為67.23 KJ/mol,是反應(yīng)通道一的速控步驟.與IM1相比較,TS1中的Rh-C(3)鍵長增大了0.098 ?,Rh原子和C(1)原子之間的距離由2.180 ?減少至1.988 ?,C(2)原子和C(3)原子的距離由之前的2.214 ?減少至2.122 ?.Rh原子和C(3)原子有相互遠離的趨勢,而Rh-C(1)以及C(2)-C(3)已經(jīng)微弱成鍵.TS1有且僅有一個虛頻為-404.49 cm-1.通過NBO分析,得到Rh-C(1) 的雙中心成鍵軌道上電子占據(jù)數(shù)為1.77815個,雙中心反鍵軌道電子占據(jù)數(shù)為0.34350個,也說明Rh-C(1) 已微弱成鍵.在中間體IM2中,C(2)-C(3)之間的鍵長和電荷密度分別為1.484 ?和0.269 a.u..C(2)與C(3)原子間的鍵長明顯縮短,電荷密度增大,C(2)-C(3)成鍵.Rh原子與C(3)原子距離增大,Rh-C(3)鍵斷開.通過TS1,實現(xiàn)了由Rh催化的芳基化交叉偶聯(lián)反應(yīng)過程.接著IM2經(jīng)過一個過渡態(tài)TS2生成中間體IM3,這是一個Rh原子轉(zhuǎn)移促使C-C偶聯(lián)發(fā)生的過程.IM2至TS2的反應(yīng)活化能為64.93 KJ/mol.TS2的虛頻唯一,值為-245.04 cm-1,說明了此過渡態(tài)的真實存在.與IM2相比較,TS2中的C(1)原子與C(4)原子的鍵長由2.753 ?縮短至1.484 ?,且存在一個0.296 a.u.的鍵鞍點電荷密度,說明C(1)-C(4)已逐漸成鍵,達到C-C偶聯(lián)的效果.而通過對TS2的NBO分析,我們可以發(fā)現(xiàn)C(1)-C(4)的雙中心成鍵軌道上電子占據(jù)數(shù)為1.96634個,雙中心反鍵軌道電子占據(jù)數(shù)為0.04119個,也說明C(1)-C(4)已微弱成鍵.從TS2到IM3,體系能量降低了128.48 KJ/mol.與TS2相比較,C(1)-C(4)鍵長更短,成鍵更穩(wěn)定;Rh原子與C(2)原子之間的距離由3.022 ?縮短至2.116 ?,逐漸成鍵.也就是說在C(1)與C(4)原子不斷靠攏的同時,Rh與I原子不斷朝C(2)方向靠近.通過對IM3的NBO分析,C(1)-C(4)鍵主要是SP-SP雜化軌道形成的σ鍵,電子占據(jù)數(shù)為1.96511個,由金屬Rh催化的C(1)-C(4)偶聯(lián)過程完成.
表1 反應(yīng)各駐點的能量G(a.u.)、相對能量Grel(KJ·mol-1)及各過渡態(tài)頻率v(cm-1)
Table 1 The energies (E/a.u.), relative energies (Erel/KJ·mol-1) and frequencies (v/cm-1) of the compounds
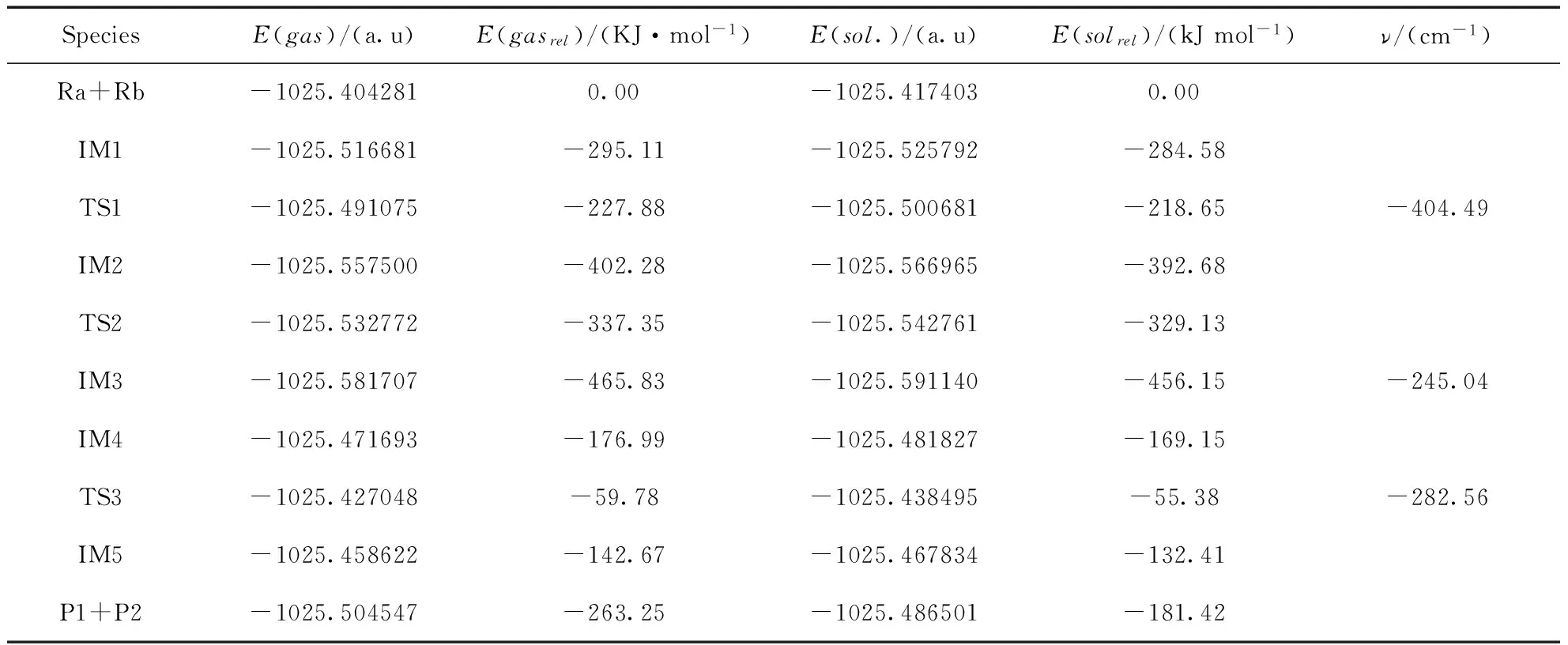
SpeciesE(gas)/(a.u)E(gasrel)/(KJ·mol-1)E(sol.)/(a.u)E(solrel)/(kJmol-1)ν/(cm-1)Ra+Rb-1025.4042810.00-1025.4174030.00IM1-1025.516681-295.11-1025.525792-284.58TS1-1025.491075-227.88-1025.500681-218.65-404.49IM2-1025.557500-402.28-1025.566965-392.68TS2-1025.532772-337.35-1025.542761-329.13IM3-1025.581707-465.83-1025.591140-456.15-245.04IM4-1025.471693-176.99-1025.481827-169.15TS3-1025.427048-59.78-1025.438495-55.38-282.56IM5-1025.458622-142.67-1025.467834-132.41P1+P2-1025.504547-263.25-1025.486501-181.42
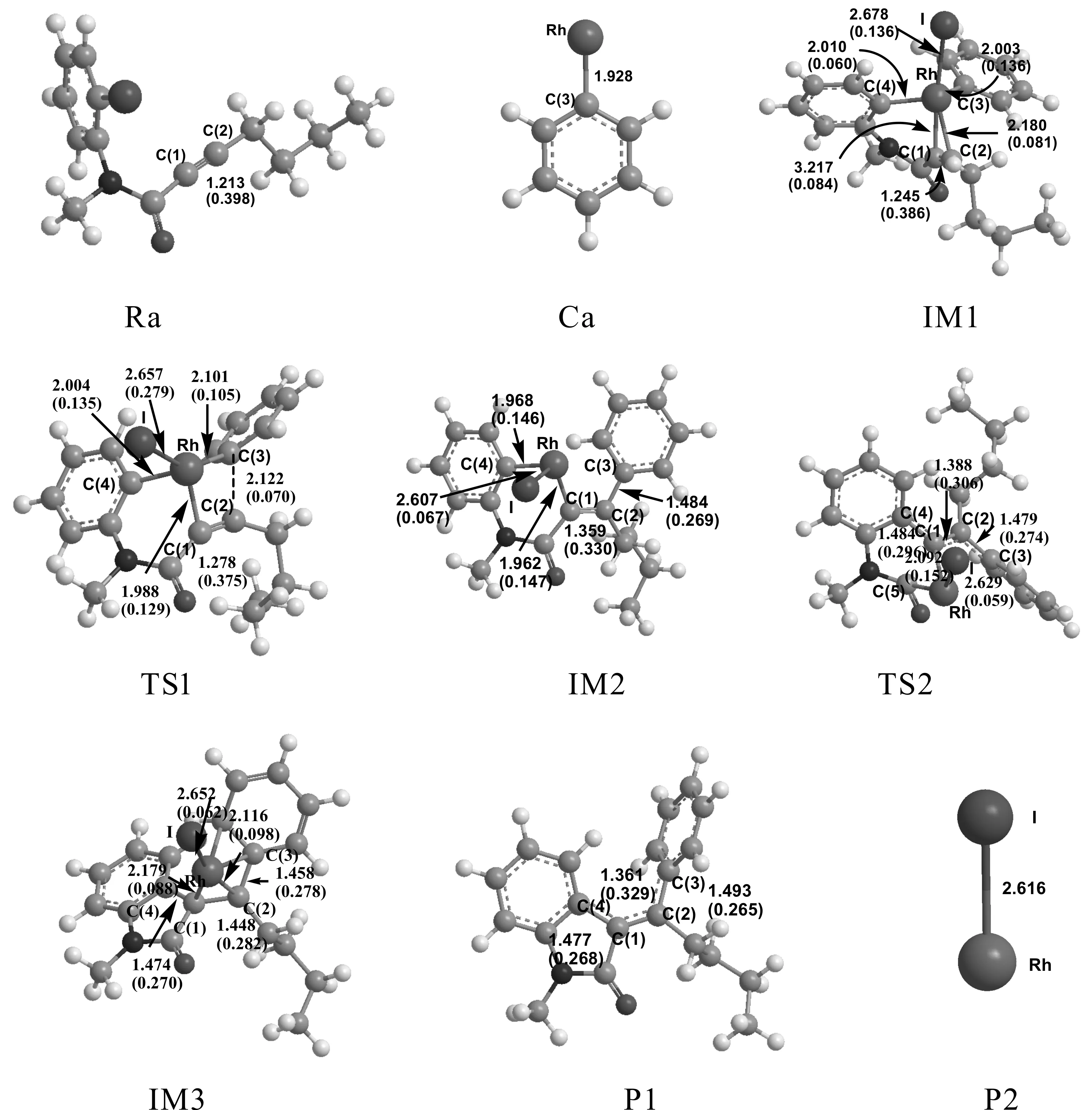
圖1 反應(yīng)通道一中涉及的化合物的幾何優(yōu)化構(gòu)型:鍵長單位為?,電荷密度單位為a.u.Fig. 1 Optimized structures of the compounds of the channel 1:bond length in angstrom units, and charge density at the bond-forming critical point in atomic units

Scheme 1 反應(yīng)通道一的微觀流程圖Scheme 1 The mechanism of the reaction channel 1

圖2 反應(yīng)通道一的能級圖Fig. 2 Schematic of energy levels in the channel 1 of the reaction
接下來IM3脫去催化劑Rh,生成產(chǎn)物P1和P2,完成整個催化過程,開始新一輪的催化循環(huán).產(chǎn)物P1中C(1)-C(4)之間的鍵長為1.477 ?,電荷密度為0.268 a.u.; C(1)-C(2)鍵長為1.361 ?,電荷密度為0.329 a.u.;C(2)-C(3)之間的鍵長和電荷密度分別為1.493 ?和0.265 a.u..最終,實現(xiàn)了產(chǎn)物的芳基化交叉偶聯(lián)以及C-C偶聯(lián)反應(yīng)過程.與實驗結(jié)果相一致.
Rh(Ⅰ)催化下的Rh芳基化合物與含氮芳基鹵代物偶聯(lián)反應(yīng)的催化循環(huán)過程一可能為:Ra+Ca →IM1 →TS1 →IM2 →TS2 →IM3→ P1+P2.反應(yīng)通道一的速控步驟為IM1 →TS1 →IM2,反應(yīng)活化能為67.23 KJ/mol.結(jié)合表1所列在相同基組水平上溶劑化條件下理論計算所得的能量數(shù)據(jù),可知能量變化趨勢和氣相條件下是一致的.
3.2 反應(yīng)通道二
因為Rh配合物容易與各種不飽和鍵發(fā)生加成,因此第二個可能存在的反應(yīng)通道初始時是由Rh的芳基化合物直接插入反應(yīng)物中不飽和的C-C鍵,反應(yīng)具體流程圖見Scheme 2.
即反應(yīng)物Ra與Rh化合物Ca發(fā)生反應(yīng)后生成中間體IM4,體系能量降低284.58 KJ/mol.說明IM4的構(gòu)型穩(wěn)定存在,插入催化劑過程較容易完成.催化劑的插入過程完成,生成一個很穩(wěn)定的Rh的烯基化合物.
接著中間體IM4經(jīng)由過渡態(tài)TS3生成另一個穩(wěn)定的中間體IM5,這是一個分子內(nèi)的氧化加成反應(yīng)過程.IM4至TS3的反應(yīng)活化能為117.21 KJ/mol,是反應(yīng)通道二的速控步驟.由過渡態(tài)TS3完成苯基的交叉偶聯(lián)到穩(wěn)定狀態(tài)IM5,體系能量降低了82.89 KJ/mol.TS3的虛頻唯一,值為-282.56 cm-1,說明了此過渡態(tài)的真實存在.與IM4相比較,C(2)原子與C(3)原子的鍵長縮短,存在一個0.113a.u.的鍵鞍點電荷密度,說明C(2)-C(3)已逐漸偶聯(lián).對TS3的NBO分析發(fā)現(xiàn)C(2)-C(3)的雙中心成鍵軌道上電子占據(jù)數(shù)為1.73685個,雙中心反鍵軌道電子占據(jù)數(shù)為0.25796個,該鍵相對能量為-0.46351 a.u.,也說明C(2)-C(3)已有成鍵趨勢.IM5與TS3相比較,C(2)-C(3)鍵的鍵長更短,成鍵更穩(wěn)定,說明IM5是一個穩(wěn)定存在的中間體.在Rh催化作用下,電子的躍遷和遷移導致苯環(huán)靠攏向炔基的另一端,完成芳基化的交叉偶聯(lián)反應(yīng)過程.通過對IM5的NBO分析,C(2)-C(3)鍵主要是SP-SP雜化軌道形成的σ鍵,雙中心成鍵軌道上電子占據(jù)數(shù)為1.96460個,雙中心反鍵軌道電子占據(jù)數(shù)為0.04065個,該鍵相對能量為-0.62967 a.u..C(2)-C(3)穩(wěn)定成鍵,Rh-C(3)斷開,由金屬Rh催化的C(2)-C(3)偶聯(lián)過程完成.接下來的反應(yīng)步驟與反應(yīng)通道一反應(yīng)步驟趨于相同,IM5通過一個分子內(nèi)的插入反應(yīng)形成IM2,Rh原子直接插入鹵素與C原子之間形成一個新的Rh化合物.系統(tǒng)能量降低了259.61 KJ/mol,反應(yīng)較易進行.IM2經(jīng)過TS2生成IM3,IM3脫去催化劑Rh,生成產(chǎn)物P1和P2,完成催化過程,開始新一輪的催化循環(huán).最終實現(xiàn)了產(chǎn)物的芳基化交叉偶聯(lián)以及C-C偶聯(lián)反應(yīng)過程.與實驗結(jié)果相一致.Rh(Ⅰ)催化下的Rh芳基化合物與含氮芳基鹵代物偶聯(lián)反應(yīng)的催化循環(huán)過程二可能為:Ra+Ca →IM4 →TS3 →IM5→IM2 →TS2 →IM3→ P1+P2.整個反應(yīng)的速控步驟為IM4 →TS3 →IM5,反應(yīng)活化能為117.21 KJ/mol.結(jié)合表1所列在相同基組水平上溶劑化條件下理論計算所得的能量數(shù)據(jù),可知能量變化趨勢和氣相條件下是一致的.與反應(yīng)通道二相比較,通道一的活化能更低,更容易發(fā)生,更容易清楚地體現(xiàn)整個偶聯(lián)反應(yīng)的微觀過程.

圖3 反應(yīng)通道二中涉及的化合物的幾何優(yōu)化構(gòu)型:鍵長單位為?,電荷密度單位為a.u.Fig. 3 Optimized structures of the compounds of the channel 2:bond length in angstrom units, and charge density at the bond-forming critical point in atomic units

Scheme 2 反應(yīng)通道二的微觀流程圖Scheme 2 The mechanism of the reaction channel 2

圖4 反應(yīng)通道二的能級圖Fig. 4 Schematic of energy levels in the channel 2 of the reaction
4 結(jié) 論
本文采用密度泛函理論(DFT)的B3LYP方法,在6-31+G(d) 基組水平上(Rh和I采用了贗勢基組LanL2DZ)研究了Rh芳基化合物與含氮芳基鹵代物偶聯(lián)反應(yīng)的催化循環(huán)過程中的兩種可能存在的反應(yīng)通道的微觀反應(yīng)機理.同時使用自洽反應(yīng)場(SCRF)極化連續(xù)模型(IEF-PCM)模擬實驗所使用的二氧六環(huán)(dioxane)溶劑效應(yīng), 采用6-311++G(d,p)基組水平對反應(yīng)機理中各化合物均進行了全參數(shù)優(yōu)化,并使用相同基組計算了氣相條件下的單點能.全文分析了此偶聯(lián)反應(yīng)的兩種可能存在的反應(yīng)通道,結(jié)論如下:(1)Rh(Ⅰ)在偶聯(lián)反應(yīng)中是一種有效的催化劑.(2)Rh配合物作為催化劑的偶聯(lián)反應(yīng)依然和其他的偶聯(lián)反應(yīng)一樣遵循氧化加成—金屬配體轉(zhuǎn)移—還原消除,催化劑進入下一個循環(huán)這樣的合成步驟.(3)在溶劑化條件下反應(yīng)能量變化趨勢和氣相條件下是一致的.(4)計算模擬出的微觀機理過程及所得結(jié)論與Ryo Shintani等的實驗推斷過程及實驗結(jié)論相符合.
[1] Zhang W W, Dai Z Q, Zhang Z Y,etal. Progress in transition-metal-catalyzed cross-coupling reaction of Grignard reagent with halides [J].ChemicalWorld, 2011, 5: 314 (in Chinese) [張煒煒, 戴志群, 張智勇, 等. 過渡金屬催化下的格氏試劑與鹵代烴的偶聯(lián)反應(yīng)研究進展[J]. 化學世界, 2011, 5: 314]
[2] Corey E J, Cheng X M.Thelogicofchemicalsynthesis[M]. New York: John Wiley & Sons, 1989.
[3] Guo X W, Li Z P, Li C J. Cross-dehydrogenative-coupling(CDC)reaction [J].Process.Chem., 2010, 22: 1434 (in Chinese) [郭興偉,李志平,李朝軍. 交叉脫氫偶聯(lián)反應(yīng)[J]. 化學進展, 2010, 22: 1434]
[4] Fleming I.Pericyclicreactions[M]. New York: Oxford University Press, 1999.
[5] Crabtree R H.Theorganometallicchemistryoftransitionmetals,4thed[M]. New York: Wiley Interscience, 2005.
[6] Tsuji J.Transitionmetalreagentsandcatalysts:innovationsinorganicsynthesis[M]. Chichester, UK: Wiley, 2002.
[7] Novák Z, Szabó A, Répási J,etal. Sonogashira coupling of aryl halides catalyzed by palladium on charcoal [J].J.Org.Chem., 2003, 68 (8): 3327.
[8] Hanamoto T, Kobayashi T. Cross-coupling reactions of (1-fluorovinyl) methydiphenylsilane1 with aryl halides and aryl triflates [J].J.Org.Chem., 2003, 68 (16): 6354.
[9] Everson D A, Shrestha R, Weix D J. Nickel-catalyzed reductive cross-coupling of aryl halides with alkyl halides [J].J.Am.Chem.Soc., 2010, 132 (3): 920.
[10] Wager K M, Daniels M H. Palladium-catalyzed cross-coupling of benzyl thioacetates and aryl halides [J].Org.Lett., 2011, 13(15): 4052.
[11] Hossain K M, Takagi K. Novel Rh(I)-catalyzed reaction of arylzinc compounds with methyl halides [J].Chem.Lett., 1999, 28: 1241.
[12] Takahashi H, Hossin K M, Nishihara Y,etal. Synthesis of functionalized benzylsilanes from arylzinc compounds and (Iodomethyl) trimethylsilane by means of a novel Rh catalysis [J].Org.Chem., 2006, 71: 671.
[13] Takahashi H, Inagaki S, Nishihara Y,etal. Novel Rh catalysis in cross-coupling between alkyl halides and arylzinc compounds possessing ortho-COX (X = OR, NMe2, or Ph) groups [J].Org.Let., 2006, 8: 3037.
[14] Yasui H, Mizutani K, Yorimitsu H,etal. Cobalt- and rhodium-catalyzed cross-coupling reaction of allylic ethers and halides with organometallic reagents [J].TetrahedronAsy., 2006, 62: 1410.
[15] Han Y F, Yang D Q. Progress of Rhodium-catalyzed asymmetric hydrogenation [J].ChemicalIndustryTimes, 2004, 18: 1 (in Chinese) [韓英峰, 揚定喬. 銠催化不對稱氫化反應(yīng)的研究進展[J].化工時刊. 2004, 18: 1]
[16] Joel M, Hawkins T J N. Asymmetric catalysis in the pharmaceutical industry [J].AngewChem.Int.Ed., 2004, 43: 3224.
[17] Jerphagon T, Renaud J, Bruneau C. Chrial monodentate phosphorus ligands for rhodium -catalyzed asymmetric hydrogenation [J].TetrtahedronAsy., 2004, 15: 2101.
[18] Dieguez M, Pamies O, Claver C. Recent advances in Rh-catalyzed asymmetric hydroformformylation using phosphite ligands [J].TetrtahedronAsy., 2004, 15: 2113.
[19] Chen B, Dingerdissen U, Krauter J G E,etal. New developments in hydrogenation catalysis particularly in synthesis of fine and intermediate chemicals [J].AppliedCatalyst., 2005, 280: 17.
[20] Blaser H U, Pugin B, Spindler F. Progress in enantioselective catalysis assessed from an industrial point of view [J].J.Mol.Catal. A:Chem., 2005, 231: 1.
[21] Xiong X D, Wang S G. Application of Platinum-group metals in asymmetric catalysis [J].ChineseJournalofRareMetals, 2005, 29(3): 363 (in Chinese) [熊曉東, 王勝國. 鉑族金屬在不對稱催化中的應(yīng)用[J]. 稀有金屬, 2005, 29(3): 363]
[22] Shintani R, Yamagami T, Hayashi T. Rhodium-catalyzed multicomponent-coupling reactions involving a carborhodation cross-coupling sequence [J].Org.Lett., 2006, 8: 4799.
[23] Reed A E, Weinhold F, Curtiss L A,etal. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint [J].J.Chem.Rev., 1988, 88 (6): 899.
[24] Bader R W F.Atomsinmolecules.Aquantumtheory[M]. Oxford: Oxford University Press, 1990.
[25] Frisch M J, Trucks G W, Schlegel H B,etal. Gaussian 03, Rev. A7. Wallingford Gaussian, Inc., CT: 2009.
Theoretical investigation on the cross-coupling reaction mechanism of rhodium aromatic compounds with nitrogen-containing aryl halides
WANG Xiao-Lan, XU Ming-Yao, HU Qing-Qian, LI Lai-Cai
(College of Chemistry and Material Science, Sichuan Normal University, Chengdu 610066, China)
The cross-coupling reaction mechanism of rhodium aromatic compounds with nitrogen-containing aryl halides has been investigated by density functional theory (DFT). The geometries and the frequencies of reactants, intermediates, transition states, and products have been calculated at the B3LYP/6-31+G(d) level, and the LanL2DZ basis has been used as the extrabasis. The vibration analysis demonstrates that the authenticity of transition states, and the reaction processes are confirmed by the changes of charge density at bond-forming critical point analyzed by the atoms in molecules theory. In addition, the nature bond orbital has been used to discuss the bond nature and orbital interactions at the same level. Meanwhile, the single point energies of the reaction process in gas and solvent at 6-311++G(d,p) level have been individually investigated with higher precision. The results indicate that the reaction mechanism and the change trend of correspondence energy at two different levels are consistent. The result of the theory study agrees with the experimental data, it indicates that the Rh(Ⅰ) is an effective catalyst in this reaction.
Density functional theory; Aromatic compounds; Nitrogen-containing aryl halides; Rh(Ⅰ)-catalyze; Cross- coupling reaction; Reaction mechanism
103969/j.issn.1000-0364.2015.12.001
2014-11-06
四川師范大學重點資助項目
王曉嵐(1984—),女,助教,碩士,主要從事應(yīng)用量子化學研究.E-mail: 29757739@qq.com
李來才. E-mail: lilcmail@163.com
O561.4
A
1000-0364(2015)06-0903-07
