幾種N-取代-3-羥基-2-吡啶硫酮Be配合物的結構與光譜性質
2015-03-22 10:27:59黃榮誼吳根華
原子與分子物理學報 2015年6期
黃榮誼, 吳根華
(安慶師范學院化學化工學院功能配合物安徽省重點實驗室, 安慶 246011)
幾種N-取代-3-羥基-2-吡啶硫酮Be配合物的結構與光譜性質
黃榮誼, 吳根華
(安慶師范學院化學化工學院功能配合物安徽省重點實驗室, 安慶 246011)
在B3LYP/6-31+G(d)級別水平上對4種Be配合物的幾何結構進行了全優化,并探討了烷基取代基對其分子的幾何結構和電子結構等方面的影響.采用TD-B3LYP方法在同樣級別水平上研究了各配合物的電子吸收光譜,分析了光譜的變化規律.上述計算結果表明,隨烷基取代基團給電子能力的增強,前線分子軌道能級升高、能隙增大、最大吸收波長發生藍移,且最大吸收波長的躍遷類型為配體內的π→π*躍遷.
Be配合物; 電子結構; 電子光譜; 含時密度泛函理論
1 引 言
近年來,功能配合物光學材料由于具有較好的熱穩定性、較高熒光效率和易成膜性等特性已成為最有應用前景的一類光致/電致發光材料[1]. 配合物發光材料的光物理、光化學性能的研究已是世界各國科學家們的關注熱點[2].迄今為止,絕大多數配合物光致/電致發光材料(稀土發光材料除外)所包含的金屬離子多屬于第Ⅱ、第Ⅲ族.如第Ⅲ族中配位數為6的Al3+和4的B3+以及第Ⅱ族中配位數為4的Be2+和Zn2+應用較為廣泛[3]. 有機配體對配合物發光材料的光物理和光化學性能起著決定作用[4]. N-取代-3-羥基-2-吡啶硫酮是一類具有O、S雙功能配位原子的螯合配體,易于與金屬離子結合形成性質優良的功能配合物材料[5-7]. 目前,在理論上還未見含該類配體的Be的配合物的研究報道. 另外,在有機配體上引入特定給/吸電子基團,以提高分子平面性、增大其共軛程度、調節其前線分子軌道能隙、改善其電荷轉移能力被認為是開發新型配合物發光材料的行之有效方法. 因此,本文以3-羥基-2-吡啶硫酮Be配合物為基本計算模型,引入一系列的烷基取代基(如甲基、乙基和丙基),從理論上探討了取代基對其結構和光譜性質的影響,以期為實驗研究提供一定的理論信息.
2 計算模型和方法
本文運用Gaussian 09 D.01程序軟件[8],采用DFT/B3LYP方法,在6-31+g(d)基組下,對4種N-取代-3-羥基-2-吡啶硫酮Be配合物(圖1)基態結構進行全優化和振動分析,振動分析計算無虛頻. 利用AIM理論對各穩定幾何構型進行了電子密度拓撲分析.另外,采用TD-DFT/6-31+g(d) 級別方法計算了其電子吸收光譜.
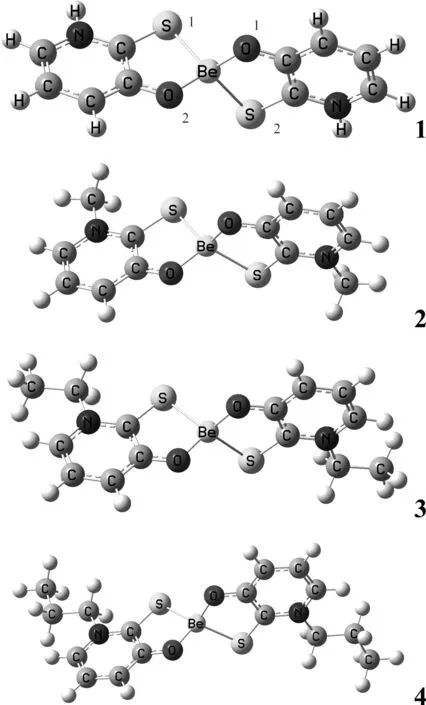
圖1 配合物1-4的分子結構模型Fig. 1 Calculation models of complexes 1-4
3 結果與討論
3.1 基態幾何構型
計算的4種配合物的穩定幾何構型如圖1所示. 分子點群均為C2群,電子態均為1A.表1給出了4種配合物的穩定構型的部分優化幾何參數. Be—O鍵鍵長在0.15825~0.15838 nm之間,Be—S鍵鍵長在0.21973~0.22110 nm之間,與文獻報道的鍵長相近. 比較4種配合物的結構參數,可以看出烷基的引入對配合物結構產生了一定的影響,烷基給電子能力的增強,使得Be—O鍵和Be—S鍵的鍵長有規律的變短,但變化幅度較小. 同時Be原子和配位原子之間的鍵角在烷基的影響下也發生了些微變化. 4種配合物的吡啶環上的C—C鍵和C—N鍵以及環外的C—O鍵、C—S鍵鍵長均介于相應鍵的單、雙鍵之間,表明吡啶環和S、O原子間共軛性較強. 因此,取代基的引入對3-羥基-2吡啶硫酮Be配合物的結構和性質將產生一定影響.
表1 在B3LYP/6-31+G(d)水平上優化的基態部分幾何參數(鍵長: ×10-1nm;鍵角: °)
Table 1 Selected geometry parameters for the ground states optimized at B3LYP/6-31G(d) level (bond length: ×10-1nm; bond angle: °)
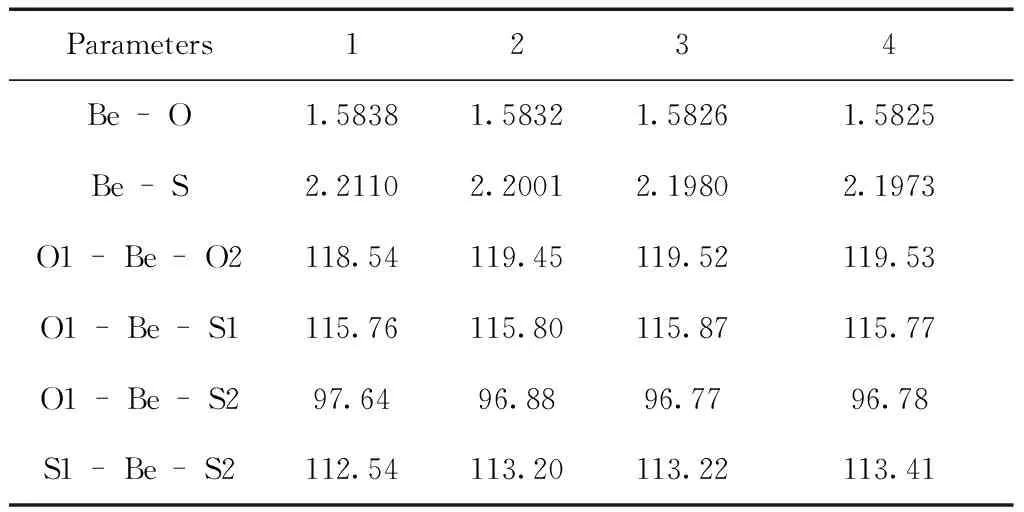
Parameters1234Be-O1.58381.58321.58261.5825Be-S2.21102.20012.19802.1973O1-Be-O2118.54119.45119.52119.53O1-Be-S1115.76115.80115.87115.77O1-Be-S297.6496.8896.7796.78S1-Be-S2112.54113.20113.22113.41
3.2 Be—O/S鍵的拓撲分析
表2給出了配位鍵Be—O/S的鍵臨界點處的電子密度拓撲參數. 結果表明4種配合物的Be—O/S的鍵臨界點的ρ(r)均較小,▽2(r)均為正值,說明在鍵臨界點處電荷是發散的,Be—O/S鍵顯示離子鍵特征,且Be—O鍵比Be—S鍵的離子性強.計算的4種配合物的橢圓度ε→0,說明Be—O/S鍵為σ配鍵. 另外,Be—S的鍵臨界點處H(r)<0,Be—O的鍵臨界點處H(r)>0,表明Be—S鍵具有部分的共價性特征,Be—O鍵共價性明顯較低. 與配合物1相比,Be—O/S鍵強度隨烷基給電子能力增大而增大,與鍵長變化的結果一致.
3.3 前線分子軌道和能級
表3和圖2給出了4種配合物分子前線軌道的組成和分布特征. 四個前線軌道中N原子上的H原子和烷基基團以及Be原子的貢獻均較少,如Ⅰ%(Be),Ⅳ%(S1)和Ⅴ%(S2)所示. 分子軌道成份主要集中在吡啶環部分和S、O原子上,如Ⅱ%(S1)和Ⅲ%(S2)所示,且均為π軌道特征. 從表3中可以看出,4種配合物的分子軌道能級分布特征極為相似,與配合物1相比,配合物2~4的前線分子軌道能級均有較小幅度的升高,且隨烷基給電子能力的增強而增大,但LUMO軌道能級升高幅度比HOMO軌道能級大,結果能隙ΔH-L變大. 配合物HOMO和LUMO能級、能隙大小順序均為4>3>2>1,與下文TD-B3LYP計算的光學帶系的變化趨勢一致. 從前線能級分布特征,可以預測配合物1的N原子上的烷基取代物的導電性會降低,其吸收和發射光譜發生藍移. 由表3和圖2所示,4種配合物的HOMO和HOMO-1之間以及LUMO和LUMO+1之間的能級差在0.0315~0.0519 eV之間,其差值較小,表明4種配合物發生HOMO→LUMO電子躍遷時,會伴隨HOMO-1→LUMO+1,且均為π→π*躍遷.
表2 配合物1-4各配位鍵臨界點處的電子密度拓撲參數(a.u.)
Table 2 Topological parameters (in a.u.)at bond critical points of complexes 1-4
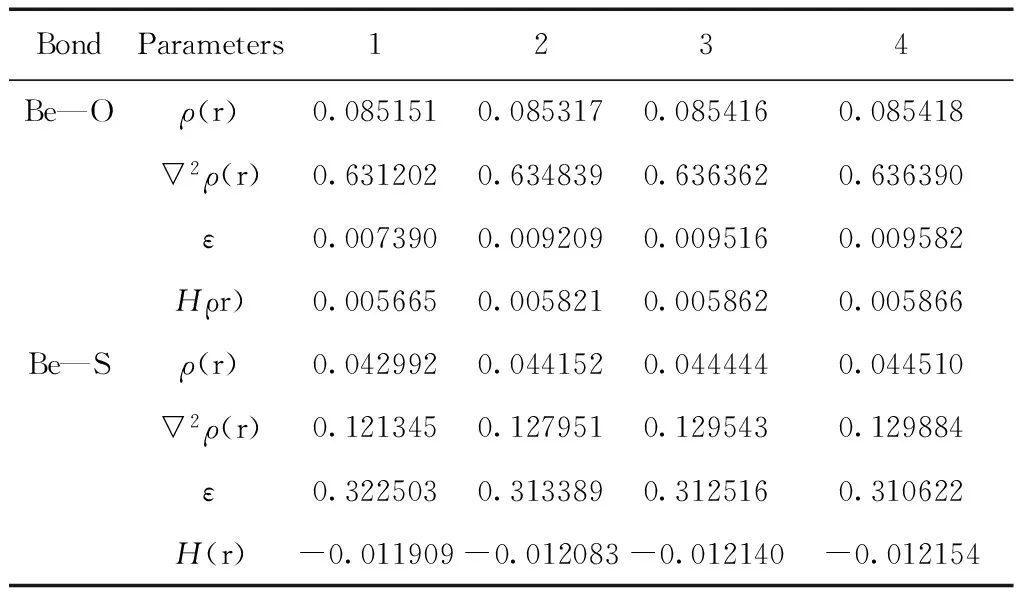
BondParameters1234Be—Oρ(r)0.0851510.0853170.0854160.085418▽2ρ(r)0.6312020.6348390.6363620.636390ε0.0073900.0092090.0095160.009582Hρr)0.0056650.0058210.0058620.005866Be—Sρ(r)0.0429920.0441520.0444440.044510▽2ρ(r)0.1213450.1279510.1295430.129884ε0.3225030.3133890.3125160.310622H(r)-0.011909-0.012083-0.012140-0.012154
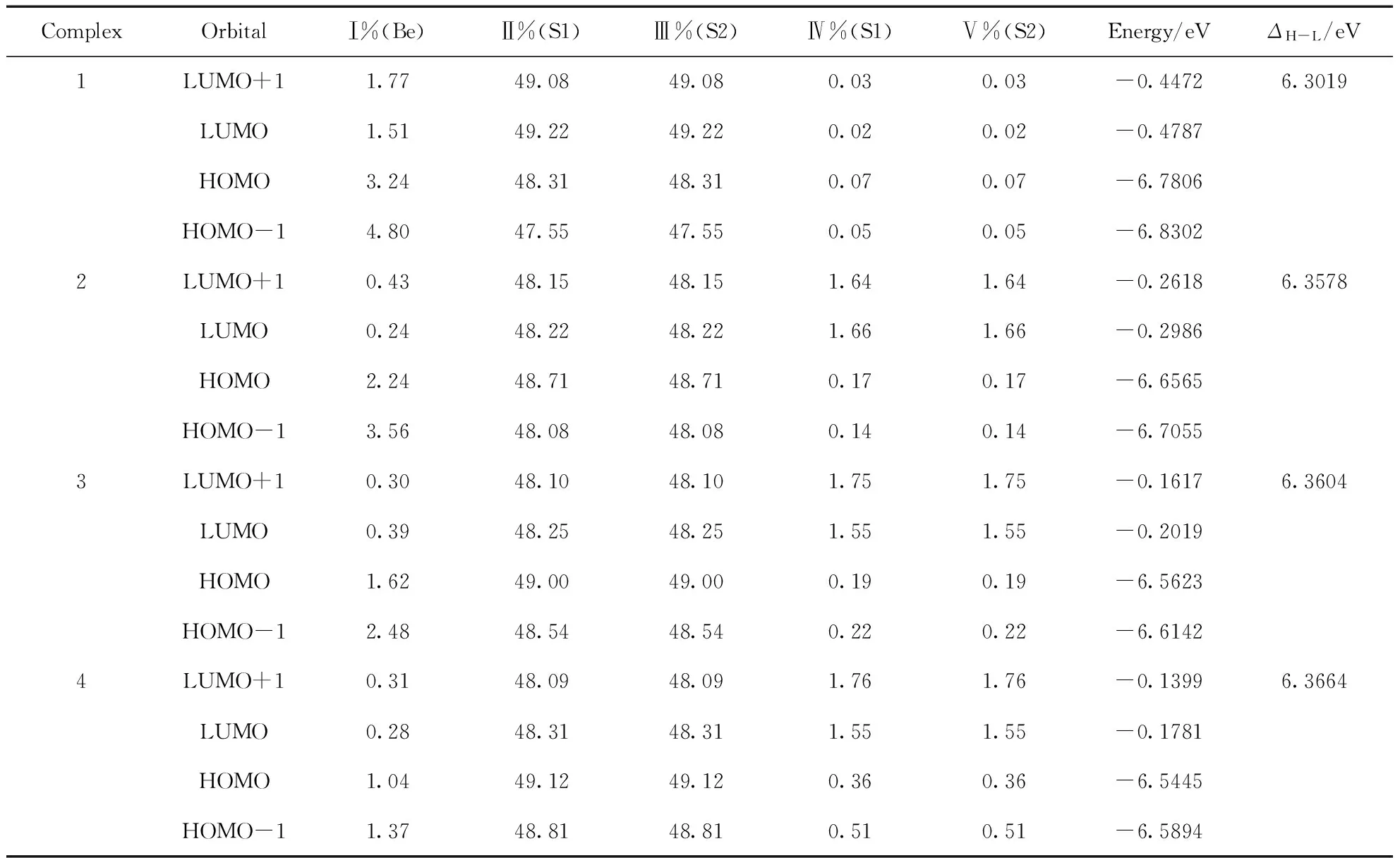
表3 配合物1-4的前線分子軌道的組成和能級及能隙

圖2 配合物1-4的前線分子軌道圖Fig. 2 The frontier molecular orbitals for complexes 1-4
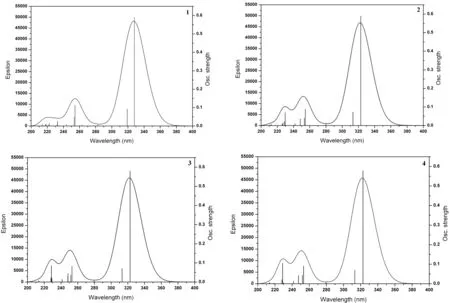
圖3 配合物1-4在氣相中模擬的吸收光譜圖Fig. 3 The simulated absorption spectra of complexes 1-4 in gas media
3.4 電子吸收光譜
在優化的基態幾何構型基礎上,利用TD-B3LYP方法計算了各配合物的電子吸收光譜.前線軌道分析表明最大吸收波長吸收峰都具有π→π*躍遷特征. 為了更好地理解烷基對配合物的電子結構和光譜性質的影響,計算了4種配合物20個單重激發態. 表4中分別列出了最大吸收波長、振子強度(f)和躍遷組成. 4種配合物的最大吸收波長分別為327.99、322.82、322.72 和322.71 nm,振子強度分別為0.5898、0.5892、0.5801和0.5778,主要是電子從HOMO-1→LUMO+1和HOMO→LUMO的躍遷,且為3-羥基-2-吡啶硫酮配體的本體π→π*躍遷,與前面分子軌道分析討論的結果一致. 圖3為模擬的4種配合物的電子吸收光譜.4種配合物吸收譜圖極為相似,在280~380 nm范圍內有一分割明顯吸收較強的單峰,在200~280 nm有兩個相互重疊的吸收較弱兩個峰. 與配合物1相比,配合物2~4的最大吸收波長都發生的較小程度的藍移,這主要是由于4種配合物具有相似的分子構型和電子結構,烷基取代基引入不會對配合物的光譜性能產生明顯的影響.
4 結 論
利用DFT和TD-DFT方法研究了4種Be配合物的電子結構和電子吸收光譜的特征. 計算結果表明4種N-取代-3-羥基-2-吡啶硫酮Be配合物中,由于烷基取代基團的修飾,分子的結構、前線軌道及能隙、和電子光譜等都受到了不同程度的影響. 與配合物1相比,其它3種取代物的能隙均增大,吸收光譜發生了藍移.理論研究結果顯示,烷基取代基對該類配合物的結構、性質與功能的影響較小. 本文研究結果可以為設計新型發光金屬配合物材料提供理論依據.
表4 配合物1-4的最大吸收波長(λ)、振子強度(f)、及軌道躍遷成份的計算結果
Table 4 The maximum absorption wavelengths (λ), oscillator strengths (f) of complexes 1-4
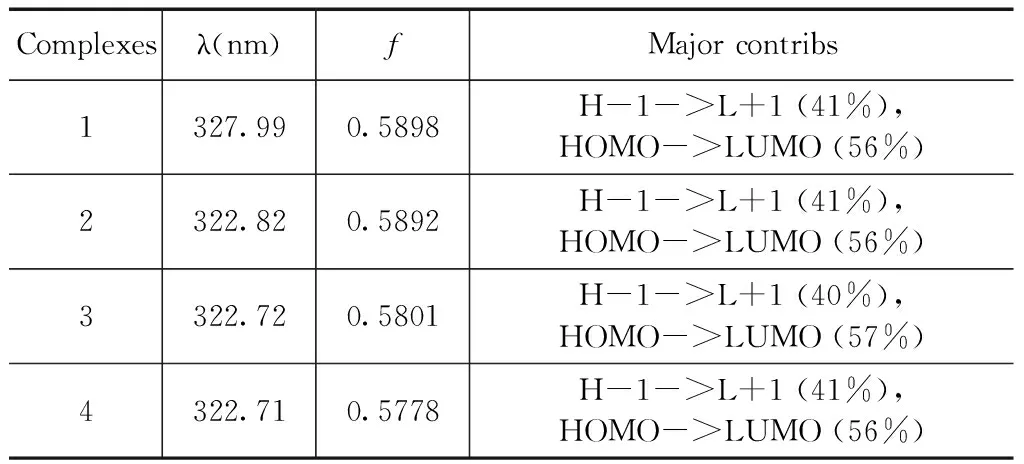
Complexesλ(nm)fMajorcontribs1327.990.5898H-1->L+1(41%),HOMO->LUMO(56%)2322.820.5892H-1->L+1(41%),HOMO->LUMO(56%)3322.720.5801H-1->L+1(40%),HOMO->LUMO(57%)4322.710.5778H-1->L+1(41%),HOMO->LUMO(56%)
[1] Evans R C, Douglas P, Winscom C J. Coordination complexes exhibiting room-temperature phosphorescence: Evaluation of their suitability as triplet emitters in organic light emitting diodes [J].Coord.Chem.Rev., 2006, 250 (15): 2093.
[2] Chi Y, Chou P T. Transition-metal phosphors with cyclometalating ligands: fundamentals and applications [J].Chem.Soc.Rev., 2010, 39 (2): 638.
[3] Wang S. Luminescence and electroluminescence of Al(Ⅲ), B(Ⅲ), Be(Ⅱ) and Zn(Ⅱ) complexes with nitrogen donors [J].Coord.Chem.Rev., 2001, 215 (1): 79.
[4] Halls M D, Schlegel H B. Molecular orbital study of the first excited state of the OLED material tris(8-hydroxyquinoline)aluminum(Ⅲ) [J].Chem.Mater., 2001, 13(8): 2632.
[5] Lewis J A, Cohen S M. Addressing lead toxicity: Complexation of lead(II) with thiopyrone and hydroxypyridinethione O,S mixed chelators [J].Inorg.Chem., 2004, 43(21): 6534.
[6] Katoh A, Yokoyama H, Matsumura Y,etal. Synthesis of metal complexes with 1-substituted 3-hydroxy-2(1H) pyridinethiones and their insulinmimetic activities [J].Heterocycles, 2010, 81(3): 585.
[7] Lewis J A, Tran B L, Puerta D T,etal. Synthesis, structure and spectroscopy of new thiopyrone and hydroxypyridinethione transition-metal complexes [J].DaltonTrans., 2005, 15: 2588.
[8] Frisch M J, Trucks G W, Schlegel H B,etal. Gaussian 09, Rev. D.01. Wallingford, CT: Gaussian, Inc., 2013.
The structures and spectrum properties of Be(Ⅱ) complexes with N-substituted 3-hydroxy-2-pyridinethione ligand
HUANG Rong-Yi, WU Gen-Hua
(Anhui Key Laboratory of Functional Coordination Compounds, School of Chemistry and Chemical Engineering, Anqing Normal University, Anqing 246011, China)
The geometry structures of four selected Be(Ⅱ) complexes were optimized at B3LYP/6-31+G(d) level of density function theory. The effects of the alkyl substituent groups on the geometrical and electronic structures have also been explored. Based on the above stable structure, the electronic absorption spectra for each complex have been theoretically investigated via TD-DFT at the aforementioned basis set level. The computational results indicate that the donation electron ability of alkyl substituent group can change the frontier molecular orbital energy levels, and results in the increases of HOMO-LUMO energy gaps. As a result, the maximum absorption spectra of the three alkyl substituent derivates have a substance blue shift accordingly.
Be(Ⅱ) coordination complex; Electronic structure; Electronic spectra; TD-DFT
103969/j.issn.1000-0364.2015.12.003
2014-10-05
國家自然科學基金(21171008);安徽高校省級優秀青年人才基金(2010SQRL108ZD)
黃榮誼(1975—),男,安徽肥西人,碩士,副教授,主要從事量子化學和功能配合物研究.E-mail: huangry@aqtc.edu.cn
O641.4
A
1000-0364(2015)06-0916-05
