第一性原理研究BiNbO4晶體的電子結構和光學性質
2015-03-23 01:04:06周朝彪冉揚強付文羽鄭興榮
原子與分子物理學報 2015年4期
周朝彪,冉揚強,吳 剛,付文羽,鄭興榮
(1.西南大學物理科學與技術學院,重慶 400715; 2.隴東學院電氣工程學院,慶陽 745000)
第一性原理研究BiNbO4晶體的電子結構和光學性質
周朝彪1,冉揚強1,吳 剛1,付文羽2,鄭興榮2
(1.西南大學物理科學與技術學院,重慶 400715; 2.隴東學院電氣工程學院,慶陽 745000)
此文用密度泛函理論的平面波贗勢方法研究BiNbO4的電子結構和光學性質.獲得了BiNbO4是一種禁帶寬度為2.74 eV的直接帶隙半導體, 價帶頂主要是由O-2p態與Bi-6s態雜化而成,而導帶底主要是由Nb-4d態構成等有益結果; 還分析得出介電函數、復折射率、能量損失等光學性質與電子態密度、能帶結構存在內在的聯系.
第一性原理; BiNbO4; 電子結構; 光學性質
1 引 言
能源短缺和環境污染是當今面臨的兩大問題,自Fujishima[1]于1972年發現TiO2在紫外光照射下催化分解水制氫以來,利用半導體光解水制氫和氧化有機物成為近年來關注的熱門課題之一[2,3].為此,尋找新型半導體光催化劑受到重視,BiNbO4是一種新型的化合物半導體材料,具有較好的導電導熱性能、耐腐蝕性和化學穩定性[4]. BiNbO4有兩種不同的結構,在高溫下合成的三斜結構(β-型)和低溫下合成的正交結構(α-型),其中正交結構有較好的光催化性能[5,6]. 目前,盡管對BiNbO4的制備與合成進行了相關的報道[7-9],但是對電子結構和光學性質的理論解釋鮮為報道.
本文利用第一性原理的廣義梯度近似方法對正交結構的BiNbO4進行了電子結構和光學性質的研究. 在結構優化的基礎上,計算了能帶結構、態密度、介電函數、吸收系數、反射系數及能量損失,并結合了電子結構對這些光學性質進行了理論分析.該研究為BiNbO4光催化材料的合成與應用提供了理論基礎和實驗指導.
2 計算方法和模型
2.1 計算方法
本文基于密度泛函理論(DFT)的第一性原理[10], 采用Materials Studio軟件中的CASTEP模塊進行計算.電子-電子之間的交換關聯勢采用GGA+PBE[11]來處理.利用周期性邊界條件,電子波函數用平面波基組展開,平面波截止能量Ecut取為340 eV. 選用超軟贗勢來描述芯電子與價電子之間的相互作用,選取價電子構型分別為:Bi-6s26p3,Nb-4d45s1和O-2s22p4. 利用Monkhorst—Pack方法[12]生成3×3×3的k點網格,保證體系能量在平面波基底水平上的收斂. 為了更好的描述電子結構的性質,我們引入了GGA+U的方法計算,對于Nb原子U=2.0 eV. 體系的結構優化采用了BFGS算法,自洽收斂精度為1.0×10-5eV/atom,原子間相互作用力收斂標準為0.03 eV/?,晶體內應力收斂標準為0.05 GPa,原子的最大位移收斂標準為1.0×10-3?.
2.2 計算模型
本文基于正交晶系[13]的BiNbO4晶體進行計算,它屬于Pnna空間群.由于考慮晶體的反演對稱性,為此用原胞內有24個原子的體系來計算和討論晶體的電子結構和光學性質.其晶格常數為a=5.682 ?,b= 11.716 ?,c=4.984 ?,以及原胞的體積為V=331.819 ?3. 如圖1所示,正交晶系的BiNbO4是由Bi層和NbO6八面體層組成的層狀結構,通過角共享方式連接了每個NbO6八面體,而Bi原子夾在兩個NbO6八面體層之間. 氧原子有兩個不等價的位置Oa位和Ob位,Oa位的氧原子與兩個Nb原子相連接,它們的鍵長分別為1.820 ?和2.149 ?,Ob位的氧原子與一個Nb原子和兩個Bi原子相連接,Ob與Nb的鍵長為2.343 ?,Ob與Bi的鍵長分別為2.121 ?和2.519 ?.
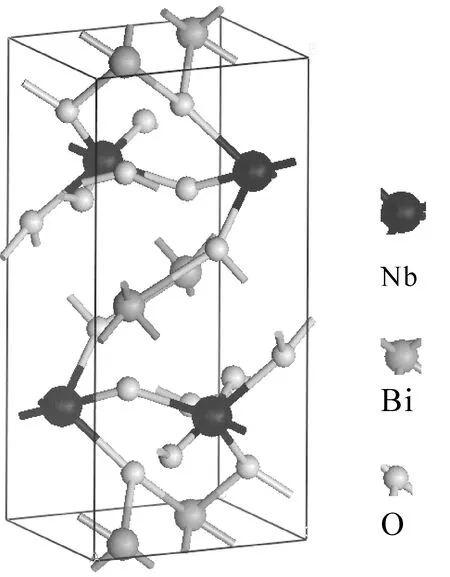
圖1 BiNbO4晶體結構Fig. 1 Crystal structures of BiNbO4
3 結果與討論
3.1 能帶結構和態密度
通過對BiNbO4能帶結構的計算,得到布里淵區高對稱點方向的能帶結構,導帶底和價帶頂都位于G點處,這說明BiNbO4為直接帶隙半導體.禁帶寬度為Eg=2.74 eV如圖2(a),計算值與實驗值Eg=2.80 eV[14]非常接近.
從態密度圖2(b)分析可知,在-18.62 eV~-15.55 eV的能量范圍內,BiNbO4的低能價帶主要是O-2s態電子組成;在-10.73 eV~-8.34 eV的能量范圍內,次低價帶主要是Bi-6s態電子組成;在-6.15 eV到費米能級范圍內,高價帶主要是O-2p態電子組成,Bi-6p態與Nb-4d態電子有少量的貢獻.BiNbO4的導帶主要是Nb-4d態所貢獻,也有少量的Bi-6p態貢獻.價帶頂主要是由O-2p態與Bi-6s態雜化而成,而導帶底主要是由Nb-4d態構成.
3.2復介電函數ε(ω)
復介電函數比宏觀光學常數更能表征材料的物理特性,更容易同物理過程的微觀模型及固體的微觀電子結構聯系起來[15].復介電函數表達式為:ε(ω)=ε1(ω)+iε2(ω)從下列公式[16]可以計算出其實部ε1(ω)與虛部ε2(ω):
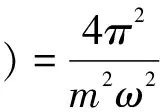
|e·MCV(K)|2×δ[EC(k)-EV(k)-?ω]}
(1)

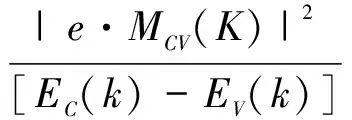
(2)


圖2 BiNbO4的能帶結構(a)和態密度(b)Fig. 2 The structures (a) and density of states (b) of the BiNbO4
從圖3(a)看出,ε1(ω)與ε2(ω)相互在曲線上升和下降沿斜率最大處出現極值,ε2(ω)共出現4個峰值,分別出現在能量為4.94 eV、12.85 eV、19.57 eV、33.97 eV,能量為4.94 eV時,ε2(ω)出現最大值,其值為8.18,當能量為36.68 eV后,ε2(ω)趨于零;靜態介電常數ε1(0)=4.94,ε1(ω)也出現四個明顯的峰值,分別出現在能量為3.10 eV、12.85 eV、19.57 eV、33.97 eV,ε1(ω)最大值為8.08,當能量在5.71~10.63 eV區間時,ε1(ω)<0,而其它區域ε1(ω)>0,且當能量大于35.76 eV后,ε1(ω)逐漸增大并趨近于0.87.
3.3 復折射率N(ω)
復折射率N(ω)=n(ω)+ik(ω),其中n(ω)為折射率,k(ω)為消光系數,從下列關系式[17,18]可以計算出n(ω)、k(ω):

(3)
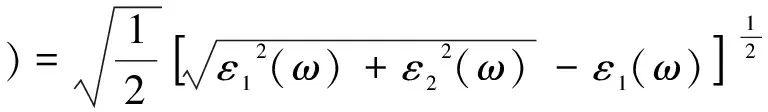
(4)
從圖3(b)中看出,靜態折射率n(0)=2.22,在低能區域,n(ω)隨著能量的增大而增大,在能量為3.26 eV時n(ω)有最大值2.90;n(ω)在3.26 eV后隨著能量的增大而急劇減小,在能量為10.96 eV時n(ω)有最小值0.01;能量10.96 eV后,n(ω)隨能量的增大而逐漸增大,在能量為13.34 eV時出現極大值0.62; 而后在13.34 eV~14.52 eV區間與19.56~21.30 eV區間及33.86~36.09 eV區間,n(ω)隨能量的增大而逐漸減小,此時n(ω)出現三極小值,分別是能量為14.52 eV對應值0.54,能量為21.30 eV對應的0.80,及能量為36.09 eV對應的0.76;能量在14.52~19.56 eV區間與21.30~33.86 eV區間,n(ω)隨能量呈遞增趨勢,n(ω)出現兩極大值,分別是能量為19.56 eV對應的0.92,能量為33.86 eV對應的1.45;在能量為36.09 eV后,n(ω)值逐漸增加并趨于0.93.消光系數k(ω)直接描述了材料電池波的衰弱,有時也稱它為阻尼常數或衰弱系數.由ε1(ω)與k(ω)的關系式ε1(ω)=n(ω)2-k(ω)2有,k(ω)出現波峰對應ε1(ω)的波谷,這由圖3也可以明顯看出;當k(ω)在能量為5.71 eV取得最大值1.88時,相應地ε1(5.71 eV)=0;能量在5.71~10.63 eV區域有k(ω)>n(ω),此時ε1(ω)<0;波谷主要出現在能量在15.45~32.95 eV區間,這與光電導率實部出現波谷的能量區域相一致,能量大于36.68 eV后有k(ω)=0,此時能量損失函數L(ω)=0.
3.4 吸收光譜α(ω)
吸收系數α(ω)可由下列關系式[17,18]計算得到:
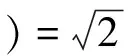
(5)
從圖4(a)看出,晶體的吸收邊大約為1.96eV,位置對應于吸收寬度,這源于價帶頂電子態向導帶底電子態的躍遷;吸收系數有四個明顯的峰值,分別在能量為7.54eV、13.77eV、20.33eV、34.95eV時出現,由態密度圖得出,這些峰主要是由O-2p、O-2s、Nb-4d、Bi-6p、Bi-6s態向導帶底的躍遷產生,它們與介電函數實部ε1(ω)出現極小值的位置(7.60 eV、14.42 eV、21.08 eV、35.76 eV)極其相近,同時還可以從圖4(a)與圖3(b)中看到吸收系數α(ω)與消光系數k(ω)的走勢及其相似,且基本上都在相同的位置取得極值.
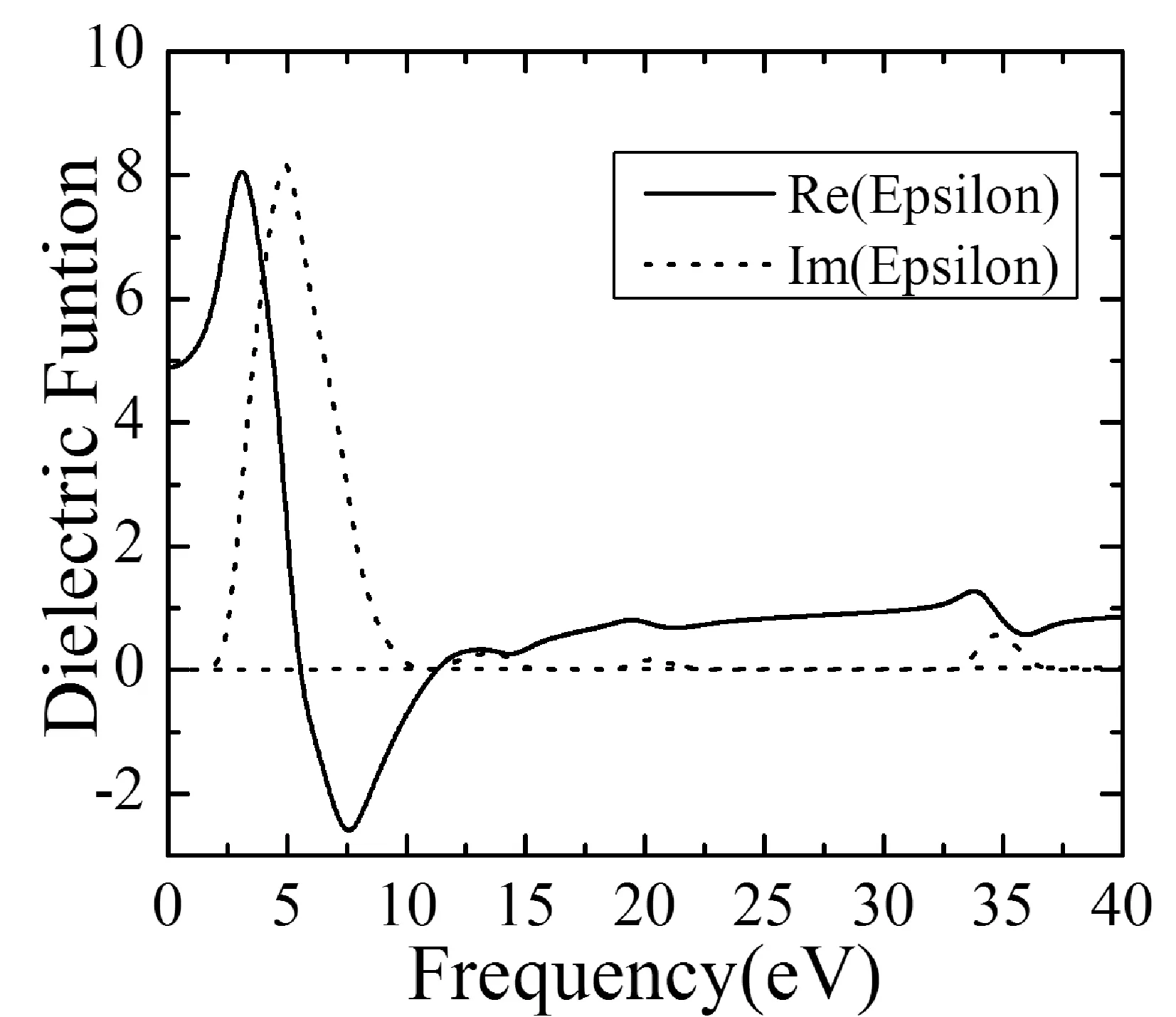
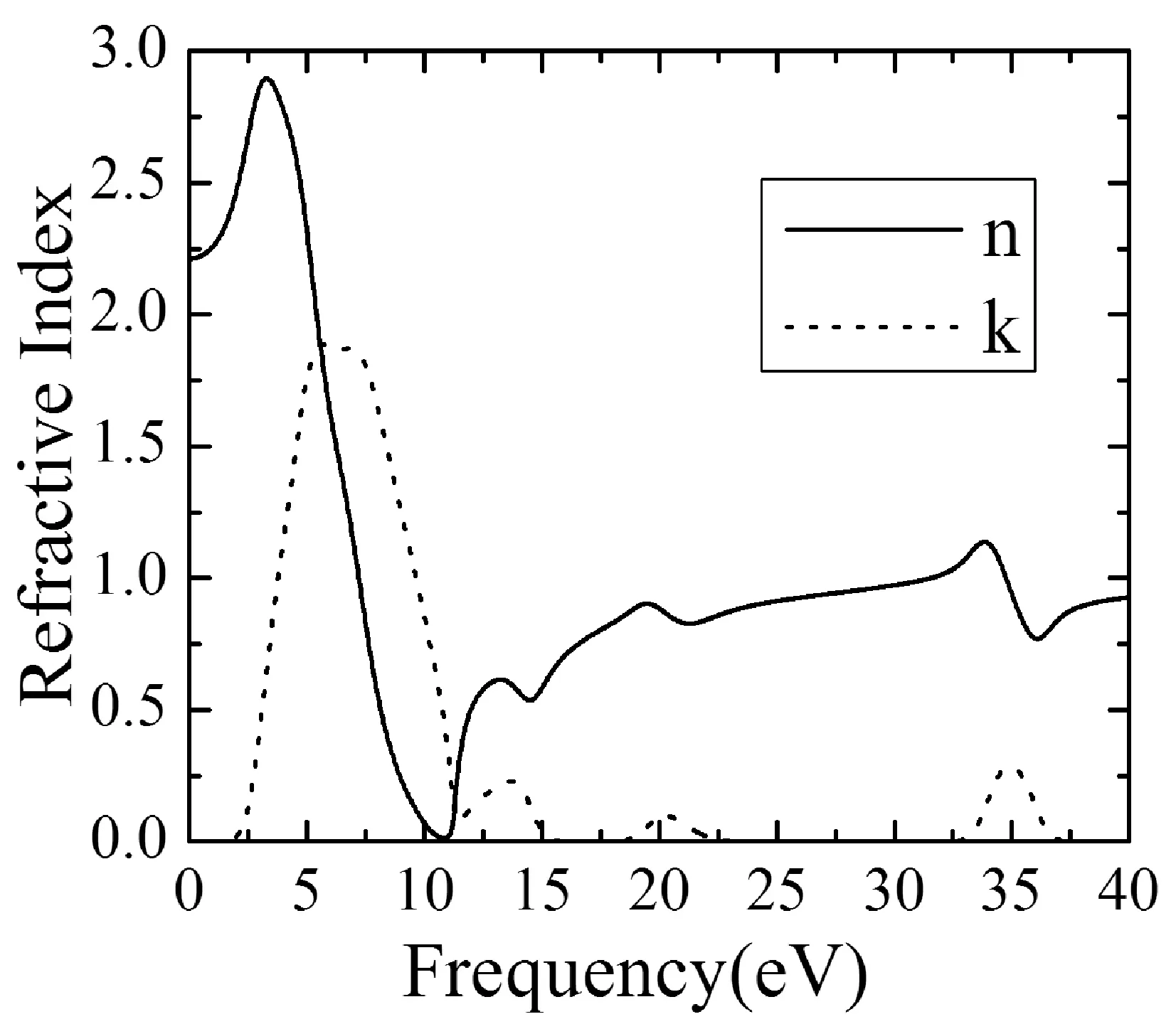
圖3 BiNbO4的復介電函數(a)和復折射率(b)Fig. 3 The complex dielectric function (a) and complex refractive index (b) of BiNbO4
3.5 反射率R(ω)
反射率R(ω)表示傳輸過程中光子的反射幾率,由反射率與介電函數的關系式[17,18]:

(6)
可計算出BiNbO4的反射率,從圖4(b)可知,反射率共出現四個峰值,分別在能量為10.79eV、14.36eV、20.97eV、35.71eV時出現,能量為10.79eV反射率有最大值0.96,峰值分別與不同軌道間的躍遷相對應,反射譜主要分布在能量為0~18.08eV間.

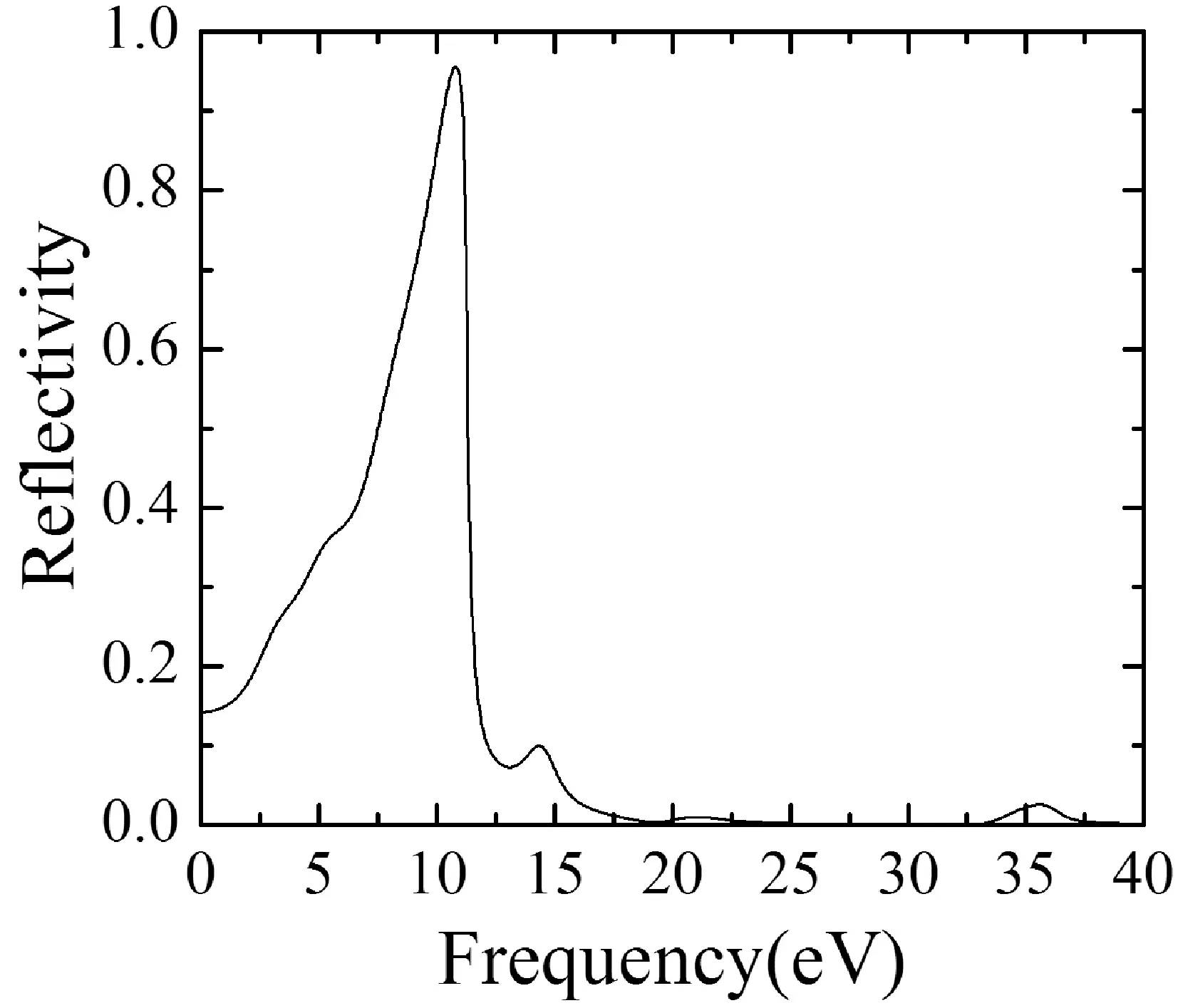
圖4 BiNbO4 的吸收率(a)和反射率(b)Fig. 4 The absorption coefficient (a) and reflectivity (b) of BiNbO4
3.6 光電導率σ(ω)
由下面光電導率公式[19]可計算得BiNbO4的光電導率值
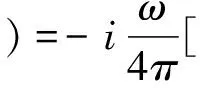
(7)
從圖5(a)可看出, 光電導率在能量為2.13eV時開始增加, 實部σ1(ω)出現四個明顯的峰值,分別在能量為5.22 eV、13.32 eV、20.27 eV、34.78 eV處產生,均由相應的帶間激發躍遷引起.σ1(ω)在能量為5.22 eV時有最大值;波谷主要出現在能量為15.49~33.02 eV的區間,這與消光系數k(ω)出現波谷的區域(15.49~33.02 eV)是一致的,在能量為10.90 eV時實部σ1(ω)出現最小值.虛部σ2(ω)在實部σ1(ω)下降沿出現極四個極大值,分別在光子能量為7.82 eV、14.52 eV、21.35 eV、35.98 eV產生;同時也有虛部σ2(ω)在實部σ1(ω)上升沿出現四個極小值, 它們分別出現在能量為3.54 eV、12.80 eV、19.51 eV、33.81 eV時.通過計算可知σ1(ω)的峰值的位置與ε2(ω)出現峰值的位置(4.94 eV、12.85 eV、19.57 eV、33.97 eV)極其相近.
3.7 能量損失函數L(ω)
能量損失函數是描述材料中快電子經過測量時能量損失的一種重要方式,從下面損失函數與介電函數關系[17,18]:
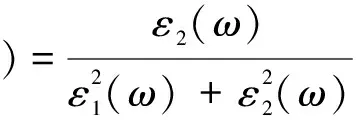
(8)
可計算出BiNO4的能量損失值,L(ω)的峰值與等離子體震蕩相關聯,相應的頻率稱為等離子體頻率.從圖5(b)中可看出,L(ω)也有四個明顯的峰值,分別在能量為11.22 eV、14.21 eV、20.49 eV、35.76 eV處產生,它們源于電子從價帶向導帶空軌道的躍遷.BiNbO4能量損失函數在能量為11.22 eV時取得最大損失值36.21.又能量大于36.68 eV后,L(ω)=0,此時對應于k(ω)=0.

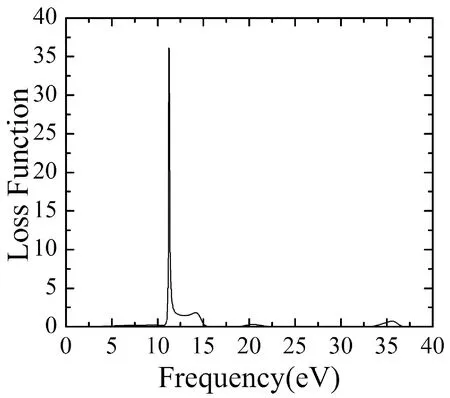
圖5 BiNbO4的光電導率(a)和損失函數(b)Fig. 5 The conductivity (a) and loss function (b) of BiNO4
4 結 論
本文基于密度泛函理論的平面波贗勢方法研究了晶體BiNbO4的能帶結構、態密度及光學性質.該研究結果表明BiNbO4是一種直接帶隙半導體,其禁帶寬度為2.74 eV,價帶頂主要是由O-2p態與Bi-6s態雜化而成,導帶底主要是由Nb-4d態構成.光學性質分析得出介電函數、復折射率、吸收光譜、能量損失等譜線的峰值是由電子態從價帶頂向導帶底的躍遷產生,這表明光學性質與電子態密度、晶體能帶結構存在內在的聯系.BiNbO4晶體的靜態介電函數為ε1(0)=4.94,吸收邊能量在1.96 eV左右,靜態折射率n(0)=2.22,BiNbO4能量損失函數在11.22 eV時取得最大損失值36.21.該研究結果為BiNbO4光催化材料的合成與應用提供較好的理論依據.
[1] Fujishima A,Honda K. Electrochemical photolysis of water at a semiconductor electrode [J].Nature,1972,238(5358): 37.
[2] Turner J A.A realizable renewable energy future [J].Science,1999,285(5428): 687.
[3] Zou Z,Ye J,Sayama K,Arakawa H. Direct splitting of water under visible light irradiation with an oxide semiconductor photocatalyst[J].Nature,2001,414: 625.
[4] Cho S Y,Youn H J,Kim D W,etal. Interaction of BiNbO4-based low-firing ceramics with silver electrodes [J].J.Am.Ceram.Soc.,1998, 81(11): 3038.
[5] Muktha B,Darriet J,Madras G,etal. Crystal structures and photocatalysis of the triclinic polymorphs of BiNbO4and BiTaO4[J].J.Solid.State.Chem.,2006,179(12): 391.
[6] Zou Z,Ye J,Arakawa H. Photocatalytic water splitting into H2and/or O2under UV and visible light irradiation with a semiconductor photocatalyst [J].Int.J.HydrogenEnergy,2003,28(6): 663.
[7] Radha R,Gupta U N,Samuel V,etal. A co-precipitation technique to prepare BiNbO4Powders [J].CeramicsInternational,2008,34(6): 1565.
[8] Radha R,Muthurajan H,Rao N K,etal. Low temperature synthesis and characterization of BiNbO4powders of BiNbO4powders [J].MaterCharact,2008,59(8): 1083.
[9] Hai Z,Ai L,Ji K,etal. Preparation and visible-light photocatalytic properties of BiNbO4and BiTaO4by a citrate method [J].J.Solid.State.Chem.,2013,202: 6.
[10] Sham L J,Schluter M. Density-functional theory of the energy gap [J].Phys.Rev.Lett., 1983,51(20): 1888.
[11] Perdew J P,Chevary J A,Vosko S H,etal. Atoms,molecules,solids,and surfaces: applications of the generalized gradient approximation for exchange and correlation [J].Phys.Rev. B,1992,46(11): 6671.
[12] Monkhorst H J,Pack J D. Special points for Brillonin-zone integrations[J].Phys.Rev. B,1976,13(12): 5188.
[13] Zou Z,Ye J,Arakawa H. Optical and structural properties of the BiTa1-xNbxO4(0≦x≦1) compounds [J].Solid.State.Commun.,2001,119(7): 471.
[14] Dunkle S,Suslick K. Photodegradation of BiNbO4powder during photocatalytic reaction [J].J.Phys.Chem. C,2009,113(24): 10341.
[15] Guo J Y,Zhong G,He K H,etal. First-principles study on electronic structure and optical proper ties of Al and Mg doped GaN [J].ActaPhys.Sin.,2008,57(6): 3740 (in Chinese)[郭建云,鄭廣,何開華,等. Al,Mg摻雜GaN電子結構及光學性質的第一性原理研究[J]. 物理學報, 2008,57(6): 3740]
[16] Li J H,Cui Y S,Zeng X H,etal,Investigations of structural phase transition,electronic structures and optical properties in ZnS [J].ActaPhys.Sin.,2013,7: 7102 (in Chinese)[李建華,崔元順,曾華祥,等. ZnS結構相變、電子結構和光學性質的研究[J]. 物理學報,2013,62(7): 7102]
[17] Sonali S,Sinha T P,Abhijit M. Electronic structure,chemical bonding,and optical properties of paraelectric BaTiO3[J].Phys.Rev. B,2000,62(13): 8828.
[18] Sun J,Wang H T,He J L,etal. Ab initio investigations of optical properties of the high-pressure phases of ZnO [J].Phys.Rev. B,2005,71(12): 125132.
[19] Fang R C.Solid-statespectroscopy[M]. Hefei: Press of University of Science and Technology of China, 2001: 10 (in Chinese)[方容川. 固體光譜學[M]. 合肥: 中國科學技術大學出版社, 2001: 10]
Investigations of electronic structure and optical properties of BiNbO4by a first-principles
ZHOU Chao-Biao1,RAN Yang-Qiang1,WU Gang1,FU Wen-Yu2,ZHENG Xing-Rong2
(1. School of Physical Science and Technology,Southwest University,Chongqing 400715,China;2.Electrical Engineering College,Longdong University,Qingyang 745000,China)
The electronic structures and optical properties of BiNbO4have been studied by using the plane wave pseudo-potential method based on density function theory.The investigations show that BiNbO4is a semiconductor with a direct band gap of 2.74 eV; the top of valence band is mainly carried out by O-2p states and Bi-6s states; while the bottom of conduction band is mainly made up by Nb-4d states.The analysis also reveal the relation of the optical properties of BiNbO4,such as the dielectric function,refractive index and energy-loss coefficient,with its density of states and band structure.
First-principles; BiNbO4; Electronic structure; Optical properties
周朝彪(1988—),男,貴州安龍人,研究生,主要從事凝聚態理論計算及量子信息的研究.
冉揚強. E-mail: zrl66@swu.edu.cn
103969/j.issn.1000-0364.2015.08.025
O481; O482
A
1000-0364(2015)08-0669-06
投稿日期:2014-05-06
猜你喜歡
數學雜志(2021年6期)2021-11-24 11:12:00
哲學評論(2021年2期)2021-08-22 01:53:34
中學生數理化(高中版.高二數學)(2021年5期)2021-07-21 02:14:46
數學年刊A輯(中文版)(2021年1期)2021-06-09 09:31:56
中等數學(2020年6期)2020-09-21 09:32:38
中華詩詞(2019年7期)2019-11-25 01:43:04
中等數學(2019年6期)2019-08-30 03:41:46
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·七年級數學人教版(2018年4期)2018-06-28 03:26:30
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
