HPGPC法測定麥芽糖鐵糖漿分子量與分子量分布
2015-07-07 16:05:23焦旭雯梁蔚陽
中國生化藥物雜志 2015年10期
焦旭雯,梁蔚陽
(廣東省食品藥品檢驗所 生檢生化室,廣東 廣州 510180)
?
HPGPC法測定麥芽糖鐵糖漿分子量與分子量分布
焦旭雯,梁蔚陽Δ
(廣東省食品藥品檢驗所 生檢生化室,廣東 廣州 510180)
目的 建立測定麥芽糖鐵糖漿的分子量與分子量分布的方法。方法 采用高效凝膠色譜法(high performance gel permeation chromatography,HPGPC),色譜柱為PSS HEMA凝膠色譜柱,檢測器為示差折光檢測器,流動相為磷酸鹽緩沖液(pH6.8),柱溫為45 ℃,流速為0.5 mL/min。結果 3批樣品的重均分子量分布在45000~47000 Da之間,精密度、重現性好。結論 該方法簡單,重現性好,可以作為控制麥芽糖鐵糖漿中分子量與分子量分布的方法。
高效凝膠色譜法;麥芽糖鐵糖漿;分子量;分子量分布
麥芽糖鐵是一種新型的多糖鐵復合物,用麥芽糊精將鐵離子穩定地絡合在其中,控制鐵的釋放,鐵離子能與鐵轉運蛋白和鐵蛋白結合以發揮作用,并防止釋放大量的游離鐵,減少有毒氧化物的形成[1]。麥芽糖鐵糖漿由麥芽糖鐵、蔗糖、山梨醇、奶油香精及適量防腐劑組成,它能有效地提高輕到中度缺鐵性貧血患者的Hb和血清鐵蛋白濃度,其作為補鐵劑不僅有合適的絡合穩定性,對胃腸道無或甚少刺激性,而且當其釋放鐵之后,本身具有多方面的生物活性,屬于對機體有益的成分,可被吸收利用[2]。多糖是天然高分子中具有多分散性的聚合物,平均分子量及其分布寬度不僅是決定多糖理化性質及活性的基本參數之一,而且與其臨床療效及過敏反應的發生關系密切,是評價藥效、控制生產和改進產品質量的主要根據和重要指標,因此研究多糖就必須掌握其分子量與分子量分布的測定方法[3]。本文利用高效凝膠色譜法測定了麥芽糖鐵分子量及分子量分布,為麥芽糖鐵糖漿生產的質量控制提供方法和依據。
1 材料與方法
1.1 材料
1.1.1 儀器:WATERS 2690型高效液相色譜儀,410示差折光檢測器,柱溫控制系統,自動進樣系統, GPC數據分析軟件(美國Waters公司);Sartorius BP211D電子分析天平(北京賽多利斯儀器有限公司);Millipore公司超純水系統。
1.1.2 藥品與試劑:3批麥芽糖鐵糖漿(瑞士Vifor制藥公司,批號:320111、426001、427001);
葡聚糖分子量標準對照品(Shodex standard p-82 lot No:130901);其余試劑均為分析純;配制溶液所用水均為Millipore公司超純水系統制得。
1.2 方法
1.2.1 色譜條件:色譜柱:PSS HEMA Bio analytical,8 mm×300 mm,孔徑分別為1000 ?與100 ?的色譜柱2根串連;示差折光檢測器檢測池溫度:45 ℃;流動相:磷酸鹽緩沖液(pH6.8)(取磷酸氫二鈉35.8 g、磷酸二氫鈉13.8 g與疊氮化鈉1.0 g,加水溶解并稀釋至5000 mL,用磷酸調節pH值至6.8);柱溫:45 ℃;流速:0.5 mL/min;進樣量:25 μL。
1.2.2 對照品溶液的制備:分別取葡聚糖對照品p5、p10、p20、p50、p100、p200,用流動相溶解并稀釋制成每1 mL中各約含10 mg的溶液,于25 ℃靜置2~3 h,作為對照品溶液。
1.2.3 供試品溶液的制備:取麥芽糖鐵糖漿適量(按Fe計算,10 mg/ml),濾過,取續濾液作為供試品溶液。
1.2.4 葡聚糖標準曲線的繪制:分別精密量取上述葡聚糖系列對照品溶液注入液相色譜儀,記錄洗脫峰的保留時間,采用GPC軟件繪制標準曲線。
1.2.5 重現性考察:取樣品(批號:427001),依1.2.3項下方法制備供試品溶液,連續進樣6針,按1.2.1項下色譜條件測定其分子量與分子量分布。
1.2.6 樣品測定:分別取3批麥芽糖鐵糖漿,依1.2.3項下方法制備供試品溶液,按1.2.1項下色譜條件測定樣品分子量與分子量分布。
2 結果
2.1 葡聚糖標準曲線的繪制 以對照品標準分子量的對數值為縱坐標、相應色譜峰的保留時間為橫坐標進行3階線性擬合。見圖10得到回歸方程lg M=-0.0005T3+0.0321T2-0.8921T+14.791,r=0.9999。見表1,校正曲線圖見圖2。
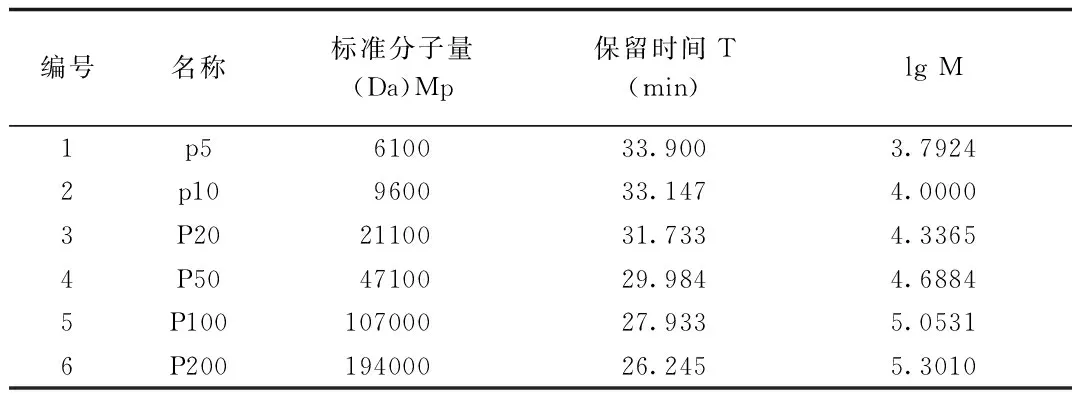
表1 葡聚糖系列對照品標準分子量及保留時間

圖1 葡聚糖系列對照品色譜圖Fig.1 A series of reference samples of the dextran
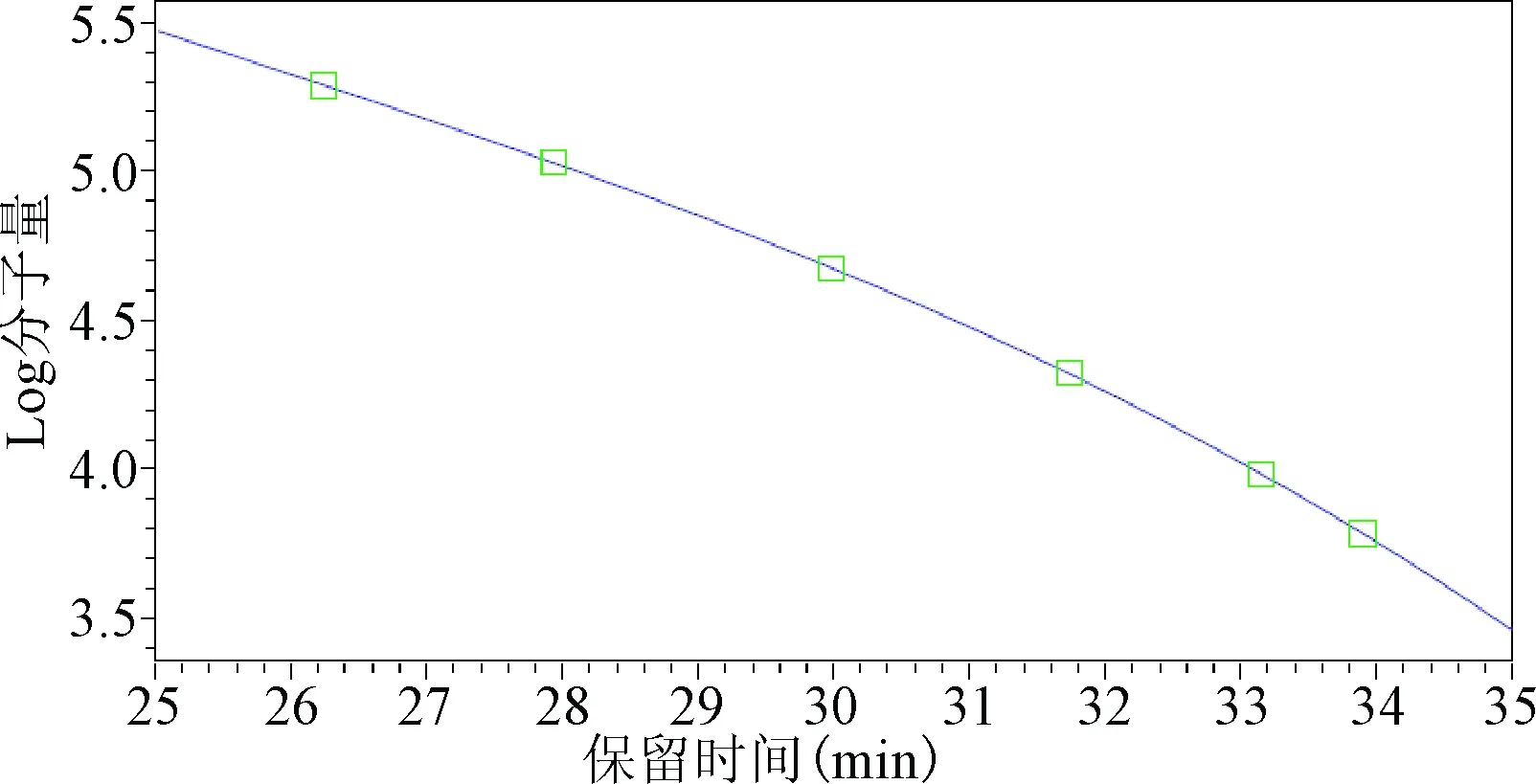
圖2 GPC校正曲線圖Fig.2 GPC calibration curve
2.2 重現性試驗 重均分子量(Mw)分別為45911、46772、46893、47056、46933、47678Da,RSD為1.21%;數均分子量(Mn)分別為31346、31780、31908、31948、31811、32489Da,RSD為1.15%。試驗結果表明建立的方法重復性良好。
2.3 樣品測定 樣品分子量與分子量分布測定結果見表2,色譜圖見圖3。同時按處方分別配制各輔料溶液,按1.2.1項下色譜條件進樣,得到各輔料峰的重疊圖譜,見圖4。
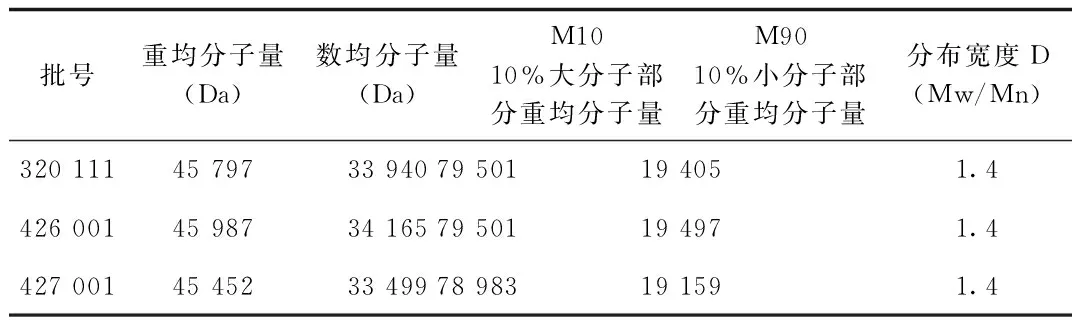
表2 3批樣品分子量與分子量分布測定結果
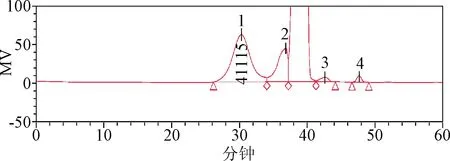
圖3 樣品色譜圖(PSS HEMA Bio analytical 1 000?、100?串聯色譜柱,批號:427001)1:麥芽糖鐵;2:山梨醇;3:奶油香精;4:防腐劑Fig.3 Sample chromatogram(PSS HEMA Bio analytical 1 000?、100? Series column No:427001)1: Iron Maltose Syrup; 2: sorbitol; 3:cream flavor;4:preservatives
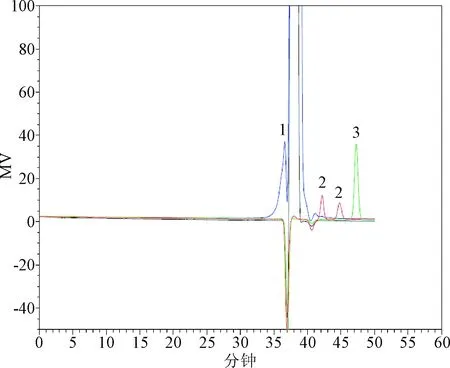
圖4 輔料重疊圖譜(PSS HEMA Bio analytical 1 000?、100?串聯色譜柱,批號:427001)1:山梨醇;2:奶油香精;3:防腐劑Fig.4 Supplementary material chromatogram(PSS HEMA Bio analytical 1 000?、100? Series column No:427001)1: sorbitol; 2:cream flavor;3:preservatives
3 討論
3.1 色譜柱的選擇 TSK-GEL PW系列色譜柱適用于以凝膠色譜的模式分析分離蛋白質、多肽、多糖、寡糖、DNA、RNA、水溶性的有機聚合物和其他水溶性大分子樣品等[4]。本研究曾嘗試選用TSK G4000PWXL凝膠色譜柱(排阻范圍:2×103~3×105)進行分析,但結果表明(見圖5),樣品基線分離效果較差,無法測得樣品10%小分子部分重均分子量。經比對,PSS HEMA Bio analytical系列色譜柱主要適用于中性和陰離子聚合物,如蛋白質、酶、多糖(羧基修飾的多糖)、水溶性淀粉等。采用孔徑為1 000?的色譜柱(排阻范圍:103~5×105)進行分析(見圖6),樣品基線分離效果有所改善,同時分子量分布寬度變窄,但仍無法測得樣品10%小分子部分重均分子量。經比較上述結果(表3),測得樣品重均分子量約為40 000~50 000,采用PSS HEMA Bio analytical,8 mm×300 mm,100?色譜柱(排阻范圍:100~6×104)進行分析,由于樣品10%大分子部分重均分子量超過排阻限,該規格色譜柱所分離的麥芽糖鐵色譜峰峰形相對較差,但可以分離出更多的輔料峰(見圖7)。綜上所述,考慮將PSS HEMA Bio analytical 1 000 ?和100 ?兩根色譜柱串聯,一方面可以得到峰形較好的麥芽糖鐵色譜峰,另一方面得到更多的輔料峰。如圖4所示,經過尺寸排阻與配位交換,麥芽糖鐵的出峰時間相應延長,山梨醇、奶油香精、防腐劑等輔料被有效分離,各組分峰基本達到基線分離[5],為獲得準確可靠的結果打下基礎。

表3 不同色譜柱測定結果比對
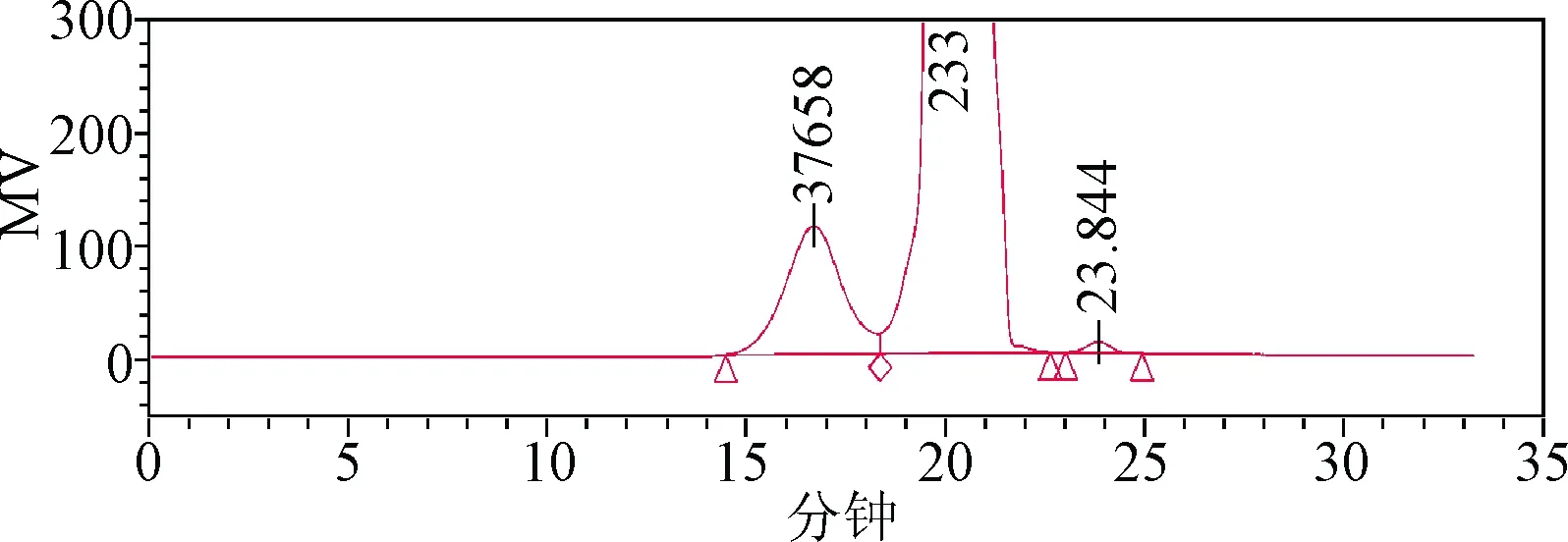
圖6 樣品色譜圖(PSS HEMA Bio analytical 1 000?色譜柱,批號:427001)Fig.6 Sample chromatogram(PSS HEMA Bio analytical 1 000? column,No:427001)
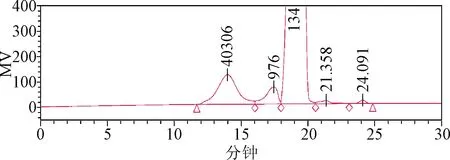
圖7 樣品色譜圖(PSS HEMA Bio analytical 100?色譜柱,批號:427001)Fig.7 Sample chromatogram(PSS HEMA Bio analytical 100? column,No:427001)
3.2 流動相的選擇 在高效凝膠色譜法中,一般使用以水為主體,具有不同pH值的多種緩沖溶液作為流動相,一般條件為pH6~8。當樣品分子帶負電荷,流動相中緩沖液濃度低時,發生靜電排斥,樣品組分靠近排阻位置被洗脫出,降低pH值既可減少樣品分子與色譜柱上的負電荷,也能降低靜電排斥[6]。同時,增大流動相離子強度可以中和電離的硅羥基與帶相同電荷的樣品分子之間的靜電排斥作用。因此本文采用磷酸鹽緩沖液(pH6.8)作為流動相,將有利于樣品洗脫,保持良好的分子量線性關系和優異的色譜峰形。
3.3 樣品濃度 由于高分子量的樣品溶液黏度較大,HPGPC的載樣容量與交互作用色譜相當。樣品的黏度較大會因為待測分子的局部濃度過高而導致峰形扭曲和變寬,此外載樣量過大也會導致這種結果[7]。本文取樣品直接濾過進樣,分別考察進樣量為5、10、25、50、100 μL,經比對,當進樣量大于25 μL時,麥芽糖鐵峰的保留時間略為延遲,顯示色譜峰有變寬的傾向。因此本法設定進樣量為25 μL,在8 mm×300 mm柱中使樣品進樣量僅為0.25 mg,可以較真實的測定出樣品分子量與分子量分布。
3.4 其他 由于葡聚糖系列標準品與樣品的結構差異、凝膠柱對其可能產生的吸附作用、分子在柱上擴散,以及葡聚糖的濃度、黏度、測定溫度的微小差異均可能影響分子量的精確性[8-11]。本文采用高效凝膠色譜法測定麥芽糖鐵糖漿中分子量及分子量分布,通過一系列的驗證試驗,證明該法操作簡便,重現性好,準確可行,為科學制訂麥芽糖鐵糖漿的質量標準提供依據。
[1] Katherine A, Lyseng-Williamson, Gillian M, et al.A Review of its Use in Iron-Deficiency Anaemia[J].Drugs, 2009, 69(6):739-756.
[2] 毛凱,馬怡璇,潘紅春,等.新型靜脈補鐵劑的研究進展[J].中國新藥雜志,2015,24(6):659-663.
[3] 汪昆華,羅傳秋,周嘯.聚合物近代儀器分析(第二版)[M].北京:清華大學出版社,2000:188.
[4] 梁蔚陽.HPGPC法測定低分子右旋糖酐氨基酸注射液的右旋糖酐40分子質量和分子質量分布[J].藥物生物技術,2008,15(3):216-218.
[5] 張義平,浦媛媛,鄒青松,等.HPGPC柱串聯法測定腸膜明串珠菌發酵產物研究[J].食品科技,2013,38(10):308-312.
[6] 丁黎.藥物色譜分析[M].北京:人民衛生出版社,2008:226-230.
[7] 陳小明,唐雅妍.現代液相色譜技術導論(第3版)[M].北京:人民衛生出版社,2012:310.
[8] 國家藥典委員會.中國藥典2015年版四部[M].北京:化學工業出版社,2015:62-63.
[9] 董曉蓉.高效液相色譜法測定蔗糖鐵注射液中蔗糖鐵的分子量和分子量分布[J].中國藥業,2006,15(4):19.
[10] 宋玉娟,任麗萍,范慧紅.硫酸軟骨素鈉分子質量及其分布的測定[J].中國新藥雜志,2011,20(18):1795-1797.
[11] 萬丹晶,陳妙芬,陳鋼.右旋糖酐鐵注射液峰位相對分子質量和重均相對分子質量的HPGPC測定[J].藥物分析雜志,2007,27(5):708-710.
(編校:譚玲)
Determination of molecular weight and weight distribution in Iron Maltose Syrup by HPGPC
JIAO Xu-wen, LIANG Wei-yangΔ
(Health Inspection Biochemical Laboratory,Guangdong Institute for Food and Drug Control, Guangzhou 510180, China)
ObjectiveTo establish a method for determining the molecular weight (Mw) and molecular weight distribution of Iron Maltose Syrup.MethodsHPGPC was used; PSS HEMA was used as column.Detector was differential refraction detector.Mobile phase was phosphate buffer solution (pH6.8) at 0.5 mL/min, column temperature was 45 ℃.ResultsThe Mw of 3 batches of Iron Maltose Syrup were 45000-47000 Da with good linearity, precision and reproducibility.ConclusionThe method is simple, reliable and accepted by the specification for controlling the molecular weight and weight distribution of Iron Maltose Syrup.
HPGPC; Iron Maltose Syrup; molecular weight; molecular weight distribution
廣東省科技計劃項目(2013B0402000029)
焦旭雯,女,碩士,主管藥師,研究方向:生化藥品檢驗,E-mail:jjxxww17@163.com;梁蔚陽,通信作者,女,碩士,主任藥師,研究方向:生化藥品及生物制品質量控制工作;E-mail:wl-1023@163.com。
R917
A
1005-1678(2015)10-0131-03
