表面增強(qiáng)拉曼光譜結(jié)合乘子效應(yīng)模型對(duì)血漿和藥片中甲巰咪唑的定量檢測(cè)
2015-07-09 18:13:19胡敏陳增萍陳瑤石彩霞俞汝勤
分析化學(xué) 2015年5期
胡敏 陳增萍 陳瑤 石彩霞 俞汝勤


摘 要 將表面增強(qiáng)拉曼散射(Surface-enhanced raman scattering,SERS)技術(shù)與乘子效應(yīng)模型結(jié)合,采用2-巰基異煙酸作為內(nèi)標(biāo),以銀納米顆粒為SERS增強(qiáng)基底,對(duì)血漿和藥片樣本中甲巰咪唑的進(jìn)行準(zhǔn)確的定量分析。實(shí)驗(yàn)結(jié)果表明,本方法對(duì)血漿樣本中甲巰咪唑的平均相對(duì)預(yù)測(cè)誤差為5.1%,檢出限為32 nmol/L;對(duì)藥片中甲巰咪唑的定量結(jié)果與LC-MS/MS方法基本一致,其加標(biāo)回收率在93.3%~110.9%之間。
關(guān)鍵詞 表面增強(qiáng)拉曼光譜; 甲巰咪唑; 2-巰基異煙酸; 乘子效應(yīng)模型; 定量分析
1 引 言
甲巰咪唑?yàn)榭辜谞钕偎幬铮话阌糜诟鞣N類型的甲狀腺功能亢進(jìn)癥【1,2】的治療。但該藥物的使用不當(dāng)或者過量使用會(huì)產(chǎn)生一定的副作用,輕則出現(xiàn)粒細(xì)胞減少,脫發(fā)、皮炎等,重則可能導(dǎo)致中毒性肝損傷。因此,必須在甲巰咪唑的生產(chǎn)和使用過程進(jìn)行嚴(yán)格的定量監(jiān)控。目前,甲巰咪唑的定量分析方法有液相色譜法【3,4】、氣相色譜-質(zhì)譜法【5,6】、電化學(xué)方法【7,8】和間接流動(dòng)注射法【9】等。但是,這些方法均具有一定的局限性:需要對(duì)復(fù)雜實(shí)際樣品進(jìn)行繁瑣費(fèi)時(shí)的預(yù)處理,或者所需儀器價(jià)格昂貴且體積龐大,不適用于大量樣品的快速在線分析。因此, 建立一種新型簡(jiǎn)便、快速、靈敏的甲巰咪唑的定量方法仍具有較重要的現(xiàn)實(shí)意義。
表面增強(qiáng)拉曼散射技術(shù)(Surface-enhanced raman scattering,SERS)具有光譜特征性強(qiáng)、靈敏度高、制樣簡(jiǎn)單,以及檢測(cè)快捷等優(yōu)點(diǎn)【10~15】。但是,樣本的SERS信號(hào)強(qiáng)度不但取決于待測(cè)樣本中待測(cè)物質(zhì)的濃度,而且與SERS增強(qiáng)基底物理性質(zhì)(如納米銀或金膠體的形狀、粒徑以及聚集度等)有關(guān)。而常用SERS增強(qiáng)基底銀(金)納米溶膠的可重現(xiàn)性和穩(wěn)定性均較差,嚴(yán)重影響樣本SERS信號(hào)的重現(xiàn)性,導(dǎo)致SERS定量分析結(jié)果的精確度遠(yuǎn)遠(yuǎn)不到實(shí)際定量分析的要求。最近,本研究小組發(fā)展了一個(gè)適用于表面增強(qiáng)拉曼光譜定量分析的乘子效應(yīng)模型(Multiplicative effects model for surface-enhanced raman spectroscopy, MEMSERS)【16,17】。 MEMSERS模型能夠有效消除SERS增強(qiáng)基底物理性質(zhì)變化對(duì)SERS定量分析結(jié)果準(zhǔn)確度的影響。本研究將SERS技術(shù)與MEMSERS相結(jié)合,實(shí)現(xiàn)了血漿和藥片中甲巰咪唑含量的快速準(zhǔn)確定量分析。
2 實(shí)驗(yàn)部分
2.1 儀器與試劑
便攜式i-拉曼785H光譜儀(上海必達(dá)泰克光電科技有限公司);1290液相色譜/G6460B系列三級(jí)四極桿質(zhì)譜聯(lián)用儀(安捷倫科技有限公司);C18反相色譜柱(150 mm×2.1 mm, 3.5 μm)。
二水合檸檬酸三鈉(C6H5Na3O7·2H2O)和AgNO3(Sigma-Aldrich 試劑有限公司)。正常人血漿從雙流正龍生化制品研究室(長沙)獲得。甲巰咪唑和2-巰基異煙酸(阿拉丁公司)。甲巰咪唑藥片(北京燕京藥業(yè)有限公司)。實(shí)驗(yàn)用水均為超純水(18.2 MΩ cm),直接從艾科浦純水系統(tǒng)(艾科浦公司)中取用。所有試劑均為分析純。
2.2 SERS增強(qiáng)基底銀納米顆粒的制備
實(shí)驗(yàn)中所用玻璃器皿均在王水(HCl-HNO3, 3∶1, V/V)中浸泡8 h以上,用超純水沖洗,烘干后使用。本研究中所用銀納米溶膠是按照Lee-Meisel方法【18】制備:稱取18 mg AgNO3, 用水溶解并定容至100 mL;將此溶液轉(zhuǎn)至圓底燒瓶并加熱至沸騰后,迅速加入2 mL檸檬酸三鈉溶液(1%),保持沸騰并繼續(xù)回流1 h后停止加熱,使溶液自然冷卻至室溫,裝瓶待用。
2.3 樣品的制備
分別用超純水和乙醇溶解適量的甲巰咪唑和2-巰基異煙酸,得到5.50 mmol/L甲巰咪唑儲(chǔ)備液和0.31 mmol/L 2-巰基異煙酸儲(chǔ)備液,于4 ℃保存?zhèn)溆谩R迫?0 μL 2-巰基異煙酸儲(chǔ)備液和不同體積的甲巰咪唑儲(chǔ)備液,用水稀釋至0.50 mL,得到11個(gè)甲巰咪唑校正樣本,其中甲巰咪唑的濃度分別為0.06, 0.18, 0.37, 0.55, 0.73, 0.92, 1.10, 1.28, 1.47, 1.65和1.83 μmol/L。
移取1.00 mL正常人血漿(用LC-MS/MS沒有檢測(cè)出甲巰咪唑)于10.0 mL離心管中,用乙腈稀釋至8.0 mL,于20 ℃以10000 r/min離心20min; 吸取4.00 mL 上清液,定容至50.0 mL,備用。移取10 μL 2-巰基異煙酸儲(chǔ)備液與適量甲巰咪唑儲(chǔ)備液,用上述血漿上清液稀釋至0.5 mL,得到甲巰咪唑濃度分別為0.28, 0.46, 0.83, 1.01, 1.19, 1.38和1.74 μmol/L的血漿預(yù)測(cè)樣本(對(duì)每個(gè)甲巰咪唑濃度水平,均配制3個(gè)重復(fù)血漿樣本)。
準(zhǔn)確稱取一片甲巰咪唑藥片(0.0721 g),用水溶解并定容至50.0 mL,再以水將其稀釋50倍,得到甲巰咪唑藥片儲(chǔ)備液。如表1所示,將20 μL藥片儲(chǔ)備液與10 μL 2-巰基異煙酸儲(chǔ)備液和適量甲巰咪唑儲(chǔ)備液混合后用超純水稀釋至0.5 mL,得到藥片預(yù)測(cè)集樣本。
2.4 樣本SERS光譜的采集
將10 μL樣本、50 μL的銀納米顆粒(AgNPs)、以及2.5 μL 2 mol/L KCl混勻后,立即用毛細(xì)管取樣,使用耦合了BAC151A拉曼影像顯微鏡采樣系統(tǒng)的便攜式i-Raman 785H光譜儀采集其SERS光譜。樣本SERS光譜采集參數(shù)如下:拉曼位移范圍500~1600 cm
激光波長785 nm,激光功率300 mW,物鏡倍數(shù)20倍,曝光時(shí)間5 s。每個(gè)樣品均重復(fù)測(cè)量3次。
2.5 LC-MS/MS實(shí)驗(yàn)endprint
為了驗(yàn)證SERS 技術(shù)結(jié)合MEMSERS模型對(duì)藥片預(yù)測(cè)集樣本中甲巰咪唑含量預(yù)測(cè)結(jié)果的準(zhǔn)確性,本研究采用1290液相色譜/G6460B系列三級(jí)四極桿質(zhì)譜聯(lián)用儀對(duì)藥片預(yù)測(cè)集中的9個(gè)樣本中甲巰咪唑的含量進(jìn)行了檢測(cè)。每個(gè)樣本均重復(fù)檢測(cè)3次。具體的色譜與質(zhì)譜實(shí)驗(yàn)條件如下: C18反相色譜柱(150 mm×2.1 mm, 3.5 μm),流動(dòng)相為0.1%甲酸-甲醇(1∶1,V/V),流速0.3 mL/min,進(jìn)樣量20 μL,質(zhì)譜離子源:電噴霧離子源,檢測(cè)模式:正離子多重反應(yīng)監(jiān)測(cè)模式,母離子:m/z 115.0 (其最優(yōu)電壓值為102 V),子離子:m/z 57.2和88.0 (其最優(yōu)的碰撞電壓分別為18和15 V),掃描時(shí)間:300 ms。表1 藥片預(yù)測(cè)樣本的實(shí)驗(yàn)設(shè)計(jì)范圍內(nèi)的SERS光譜數(shù)據(jù)進(jìn)行后續(xù)的定量分析。
由于樣本的SERS光譜信號(hào)強(qiáng)度除了與待測(cè)分析物濃度有關(guān)之外,還會(huì)受SERS增強(qiáng)基底(如銀納米顆粒)物理性質(zhì)變化的影響,這使得樣本的SERS光譜信號(hào)強(qiáng)度與待測(cè)分析物的濃度之間的關(guān)系常不為簡(jiǎn)單的線性關(guān)系。本研究采用如下MEMSERS模型對(duì)甲巰咪唑樣本的表面增強(qiáng)拉曼光譜進(jìn)行定量分析。
xk=bk·(c2-MNA,k·r2-NMA+cMMI,k·rMMI)+dk k=1,2,......K(1)
其中,xk代表第k個(gè)校正樣本的SERS光譜;c2-MNA, k和cMMI, k分別代表第k個(gè)校正樣本中2-巰基異煙酸和甲巰咪唑的濃度;r2-MNA和rMMI分別代表2-巰基異煙酸和甲巰咪唑分子的拉曼散射特性;乘子參數(shù)bk代表SERS增強(qiáng)基底物理性質(zhì)等因素變化對(duì)第k個(gè)校正樣本的SERS光譜信號(hào)產(chǎn)生的乘子效應(yīng);dk代表背景干擾和SERS增強(qiáng)基底物理性質(zhì)變化對(duì)第k個(gè)校正樣本SERS光譜信號(hào)產(chǎn)生的非乘子效應(yīng)。由于內(nèi)標(biāo)物2-巰基異煙酸在每個(gè)樣本中的濃度c2-MNA, k是固定不變的,因此MEMSERS模型中校正樣本的乘子參數(shù)bk(k=1, 2, …, K)可以用改進(jìn)光程估計(jì)與糾正的方法(Modified optical path length estimation and correction, OPLECm)【17】估計(jì)出來。獲得乘子參數(shù)bk(k=1, 2, …, K)后,采用多元校正方法(如Partial least squares regression, PLSR)在xk與bk之間,以及xk與bk·cMMI,k之間建立兩個(gè)校正模型。獲得待測(cè)樣本的SERS光譜xtest后,待測(cè)樣本中甲巰咪唑的濃度可以由第二個(gè)校正模型的預(yù)測(cè)值除以第一個(gè)校正模型的預(yù)測(cè)值而獲得。
本研究將用水配制的甲巰咪唑校正集樣本的SERS光譜作為校正集,在其上建立MEMSERS和PLSR校正模型,然后將所建立的MEMSERS和PLSR校正模型用于預(yù)測(cè)血漿預(yù)測(cè)集樣本和藥片預(yù)測(cè)集樣本中甲巰咪唑的濃度。MEMSERS和PLSR校正模型中使用的最優(yōu)潛變量數(shù)均采用交互驗(yàn)證法確定。采用預(yù)測(cè)均方根誤差(RMSEP):
RMSEP=Nk=1(cMMI,k-MMI,k)2/N(2)
cMMI, k和MMI, k分別代表第k個(gè)待測(cè)樣本中甲巰咪唑的真實(shí)濃度和預(yù)測(cè)濃度;N代表待測(cè)樣本的數(shù)目)。平均相對(duì)預(yù)測(cè)誤差(ARPE):
ARPE=1NNi=1(cMMI,k-MMI,k)/cMMI,k×100%(3)
采用RMSEP和ARPE評(píng)價(jià)和比較MEMSERS與PLSR校正模型的預(yù)測(cè)結(jié)果。
3 結(jié)果與討論
3.1 內(nèi)標(biāo)濃度的優(yōu)化
采用內(nèi)標(biāo)加入法對(duì)待測(cè)樣本中的甲巰咪唑進(jìn)行SERS定量分析。內(nèi)標(biāo)物2-巰基異煙酸(其SERS特征峰數(shù)量少且信號(hào)較強(qiáng))和待測(cè)分析物2-巰基異煙酸均含有巰基,它們可通過與銀納米顆粒表面形成穩(wěn)定的AgS鍵而修飾在銀納米顆粒表面。由于甲巰咪唑與2-巰基異煙酸之間可能存在對(duì)銀納米顆粒表面上結(jié)合位點(diǎn)的競(jìng)爭(zhēng)現(xiàn)象,作為內(nèi)標(biāo)物加入的2-巰基異煙酸的濃度若太低,則其SERS信號(hào)太弱,起不到內(nèi)標(biāo)作用;但2-巰基異煙酸濃度也不能太高,否則會(huì)影響甲巰咪唑的檢測(cè)靈敏度。為了實(shí)現(xiàn)對(duì)本研究所考察濃度范圍內(nèi)的甲巰咪唑進(jìn)行較準(zhǔn)確的定量分析,將所加入的2-巰基異煙酸的濃度設(shè)置為一個(gè)比較適中的值,即6.2 μmol/L。
3.2 增強(qiáng)基底物理物質(zhì)對(duì)樣本SERS光譜信號(hào)的影響
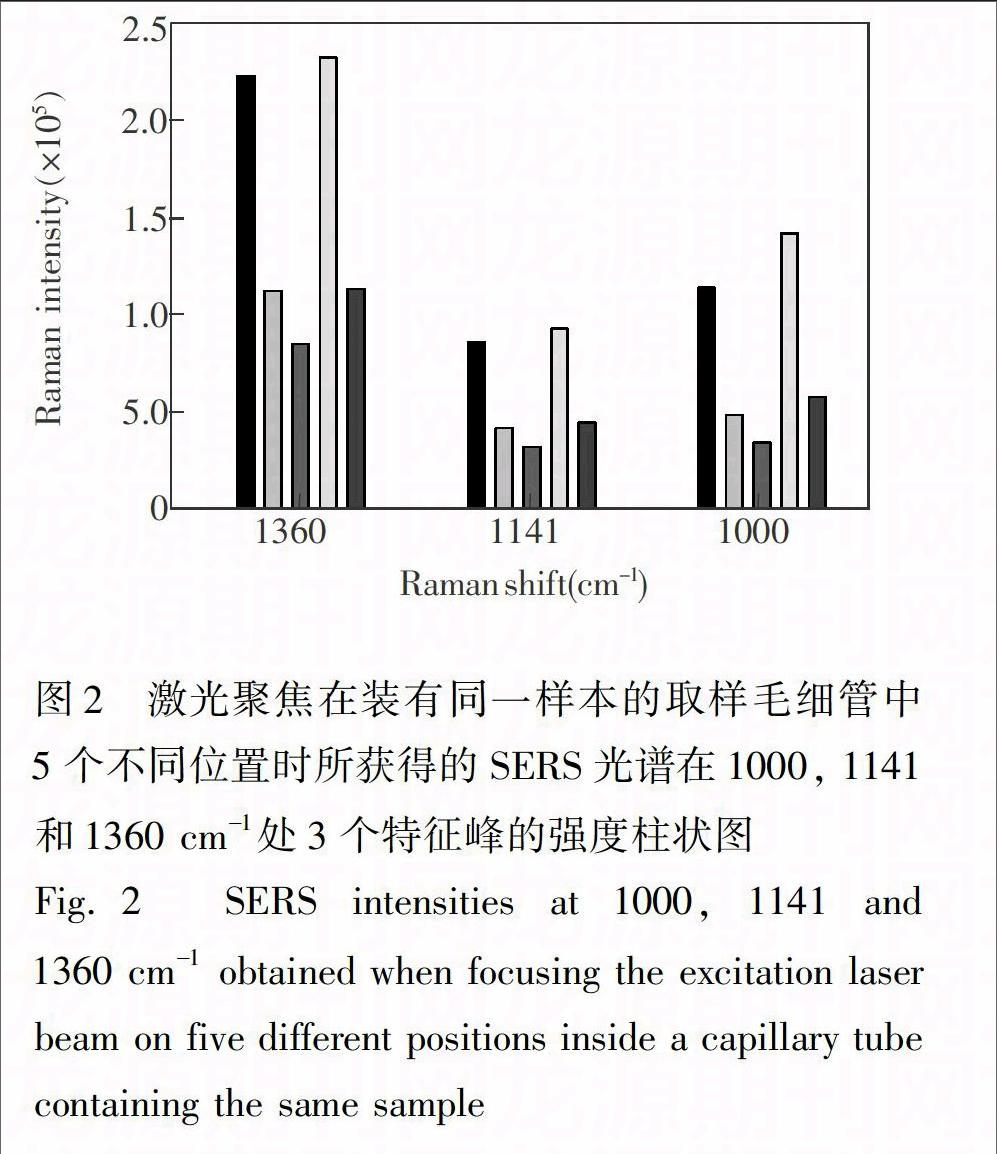
在SERS定量分析過程中遇到的最大問題是:樣本的SERS光譜信號(hào)重現(xiàn)性較差。如圖2所示,在其它實(shí)驗(yàn)條件完全一樣的情況下,將激光聚焦在同一根取樣毛細(xì)管(毛細(xì)管中裝有同一銀納米顆粒-KCl-樣本混合物)中不同位置所獲得一組SERS光譜之間存在明顯差異。這主要是由SERS增強(qiáng)基底納米銀顆粒的形狀和大小分布不均勻所致。由OPLECm所估計(jì)出來的甲巰咪唑校正集樣本的乘子參數(shù)bk在1.0~2.6范圍內(nèi)變動(dòng),表明SERS增強(qiáng)基底納米銀顆粒的形狀和大小分布不均勻性對(duì)樣本SERS光譜信號(hào)有很大影響。必須采取措施消除這種不利影響,才能實(shí)現(xiàn)對(duì)血漿預(yù)測(cè)集樣本和藥片預(yù)測(cè)集樣本中甲巰咪唑的準(zhǔn)確定量分析。
3.3 樣品分析
3.3.1 血漿樣本中甲巰咪唑的定量分析 將建立在用水配制的甲巰咪唑校正集樣本SERS光譜數(shù)據(jù)上的MEMSERS和PLSR校正模型應(yīng)用于血漿預(yù)測(cè)集樣本中甲巰咪唑濃度的定量分析。如表2所示,PLSR模型的RMSEP和ARPE值分別為0.13 μmol/L和11.0%。PLSR模型預(yù)測(cè)值與實(shí)際值之間的偏差相對(duì)較大。此外,PLSR模型對(duì)同一樣本的3次重復(fù)測(cè)量SERS光譜的定量分析結(jié)果的標(biāo)準(zhǔn)方差較大。這些現(xiàn)象表明, PLSR模型不能有效消除SERS增強(qiáng)基底物理性質(zhì)的不均一性對(duì)SERS定量分析結(jié)果的不利影響。相比之下,MEMSERS模型給出的甲巰咪唑濃度的預(yù)測(cè)值與實(shí)際值非常接近,而且其對(duì)同一樣本的3次重復(fù)測(cè)量SERS光譜的定量分析結(jié)果的標(biāo)準(zhǔn)方差也較小。MEMSERS的RMSEP和ARPE值分別為0.05 μmol/L和5.1%,均明顯小于PLSR模型的相應(yīng)值。另外,通過估算得到MEMSERS對(duì)血漿體系中甲巰咪唑的檢出限為32 nmol/L,優(yōu)于文獻(xiàn)中采用的氣相色譜-質(zhì)譜法的檢出限(45 nmol/L)【5, 6】。endprint
為了驗(yàn)證MEMSERS模型預(yù)測(cè)結(jié)果的可靠性,采用另一份正常人的血漿(新血漿),按照上述血漿預(yù)測(cè)樣本的配制方法重新配制一組血漿預(yù)測(cè)樣本,并使用建立的MEMSERS模型對(duì)這一組血漿預(yù)測(cè)樣本中的甲巰咪唑濃度進(jìn)行定量分析。結(jié)果表明,MEMSERS模型的預(yù)測(cè)結(jié)果的準(zhǔn)確度很高,其RMSEP和ARPE值分別為0.06 μmol/L和5.4%,與前面所獲得結(jié)果幾乎完全一致。因此,待測(cè)樣本基質(zhì)的變化也不影響MEMSERS對(duì)血漿預(yù)測(cè)集樣本中甲巰咪唑的定量分析結(jié)果。
3.3.2 藥片中甲巰咪唑的定量分析 SERS技術(shù)結(jié)合MEMSERS模型和LC-MS/MS對(duì)一片藥片中甲巰咪唑含量的測(cè)定值分別為5.4和5.0 mg。這兩種方法的檢測(cè)結(jié)果基本一致,并且十分接近該藥片所附說明書上標(biāo)注的規(guī)格(5 mg/片)。表3列出了MEMSERS和 LC-MS/MS對(duì)藥片預(yù)測(cè)集樣本中甲巰咪唑的定量分析結(jié)果。從表3可知,MEMSERS定量分析結(jié)果的平均加標(biāo)回收率在93.3%~110.9%之間,與LC-MS/MS定量分析結(jié)果的準(zhǔn)確度基本一致,表明SERS技術(shù)結(jié)合MEMSERS模型方法能準(zhǔn)確測(cè)定復(fù)雜體系中甲巰咪唑的含量,并且有望發(fā)展成為復(fù)雜體系中甲巰咪唑含量的常規(guī)定量分析方法。表3 MEMSERS和 LC-MS/MS對(duì)藥片預(yù)測(cè)樣本中甲巰咪唑的定量分析結(jié)果
4 結(jié) 論
采用了MEMSERS模型消除SERS增強(qiáng)基底物理性質(zhì)的不均一性對(duì)SERS定量分析結(jié)果的不利影響,以實(shí)現(xiàn)血漿和藥片樣本中甲巰咪唑的準(zhǔn)確定量檢測(cè)。結(jié)果表明,建立在超純水配制的甲巰咪唑校正集樣本SERS光譜數(shù)據(jù)上的MEMSERS模型能夠從血漿預(yù)測(cè)集樣本和藥片預(yù)測(cè)集樣本的SERS光譜中準(zhǔn)確預(yù)測(cè)出相應(yīng)甲巰咪唑的含量。MEMSERS對(duì)血漿預(yù)測(cè)集樣本中甲巰咪唑含量的平均相對(duì)預(yù)測(cè)誤差約為5.1%,檢出限達(dá)到了32 nmol/L,而且待測(cè)樣本的基質(zhì)變化不影響MEMSERS定量分析結(jié)果的準(zhǔn)確度;MEMSERS模型對(duì)藥片預(yù)測(cè)集樣本中甲巰咪唑含量的預(yù)測(cè)結(jié)果的回收率在93.3%~110.9%之間,與LC-MS/MS對(duì)照實(shí)驗(yàn)的結(jié)果基本一致。SERS技術(shù)與MEMSERS模型相結(jié)合檢測(cè)甲巰咪唑的方法具有簡(jiǎn)便、快捷、靈敏度高、檢出限低的特點(diǎn),有望發(fā)展成為復(fù)雜體系中甲巰咪唑含量的常規(guī)定量分析方法。
References
1 Weetman A P, McGregor A M, Hall R. Clin. Endocrinol., 1984, 21(2): 163-172
2 Kendall-Taylor P. Br. Med. J. 1984, 288(6416): 509
3 Hollosi L, Kettrup A, Schramm K W. J. Pharm. Biom. Anal., 2004, 36(4): 921-924
4 Kusmierek K, Bald E. Talanta, 2007, 71(5): 2121-2125
5 Zou Q H, Liu Y, Xie M X, Han J, Zhang L. Anal. Chim. Acta, 2005, 551(1): 184-191
6 Zhang L, Liu Y, Xie M X, Qiu Y M. J. Chromatogr. A, 2005, 1074(1): 1-7
7 Shahrokhian S, Ghalkhani M. Electroanalysis, 2008, 20(10): 1061-1066
8 Sun J Y, Zhang C Y, Xiao X L, Niu L, You T Y, Wang E K. Electroanalysis, 2005, 17(18): 1675-1680
9 Economou A, Tzanavaras P D, Notou M, Themelis D G. Anal. Chim. Acta, 2004, 505(1): 129-133
10 Kneipp K, Wang Y, Kneipp H, Perelman L T, Itzkan I, Dasari R R, Feld M S. Phys. Rev. Lett., 1997, 78(9): 1667
11 Nie S, Emory S R. Science, 1997, 275(5303): 1102-1106
12 LIANG Man-Fen, ZHOU Dian-Ming, WANG Yu, CHEN Cui-Hua, JIANG Jian-Hui. Chinese J. Anal. Chem., 2013, 41(9): 1341-1347
梁滿芬, 周殿明, 王 玉, 陳翠花, 蔣健暉. 分析化學(xué), 2013, 41(9): 1341-1347
13 Otto A, Mrozek I, Grabhorn H, Akemann W. J. Phys.: Condens. Matter, 1992, 4(5): 1143
14 NI Dan-Dan, WANG Wei-Wei, YAO Jian-Lin, ZHANG Xie-Jiao, GU Ren-Ao. Spectroscopy and Spectral Analysis, 2011, 31(2): 394-397
倪丹丹, 王偉偉, 姚建林, 張雪嬌, 顧仁敖. 光譜學(xué)與光譜分析, 2011, 31(2): 394-397
15 FAN Xiao-Min, ZOU Wen-Jun, GU Ren-Ao, YAO Jina-Lin. Chem. J. Chinese Universities, 2008, 29(1): 130-134
范曉敏, 鄒文君, 顧仁敖, 姚建林. 高等學(xué)校化學(xué)學(xué)報(bào), 2008, 29(1): 130-134
16 Xia T H, Chen Z P, Chen Y, Tin J W, Yu R Q. Anal. Methods, 2014, 6(7): 2363-2370
17 Song J, Chen Z P, Jin J W, Chen Y, Yu R Q. Chemom. Intell. Lab. Syst., 2014, 135: 31-36
18 Lee P C, Meisel D. J. Phys. Chem., 1982, 86(17): 3391-3395
19 Tin J W, Chen Z P, Li L M, Steponavicius R, Thennadil S N, Yang J, Yu R Q. Anal. Chem., 2011, 84(1): 320-326endprint
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
現(xiàn)代臨床醫(yī)學(xué)(2022年4期)2022-09-29 07:38:00
昆明醫(yī)科大學(xué)學(xué)報(bào)(2021年4期)2021-07-23 01:21:50
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2020年10期)2020-11-26 08:24:50
數(shù)學(xué)物理學(xué)報(bào)(2020年2期)2020-06-02 11:29:24
云南醫(yī)藥(2019年3期)2019-07-25 07:25:14
光學(xué)精密工程(2016年6期)2016-11-07 09:07:19
海南醫(yī)學(xué)(2016年8期)2016-06-08 05:43:00
核科學(xué)與工程(2015年4期)2015-09-26 11:59:03
醫(yī)學(xué)研究雜志(2015年9期)2015-07-01 17:28:15
