煤焦油加氫脫氮反應網絡及催化劑研究進展
2015-07-25 09:09:44李貴賢曹彥偉李夢晨李昱劉珍珍白宏偉
化工進展 2015年5期
關鍵詞:催化劑
李貴賢,曹彥偉,李夢晨,李昱,劉珍珍,白宏偉
(蘭州理工大學石油化工學院,甘肅 蘭州 730050)
“富煤,少氣,缺油”是我國目前的能源現狀。我國現代煤化工正快速發展,作為副產物的煤焦油產量逐年增加,2014 年我國煤焦油產量估計在2200萬噸左右且產量在逐年增加。煤焦油主要用于深加工生產一些重要的化工原料[1],其余的煤焦油被作為粗燃料油直接燃燒,由于煤焦油中含有大量的S、N 化合物,其S、N 含量是市場上機動車燃料的數十倍甚至百倍,燃燒時排放出大量的NOx和SOx污染物,給環境造成了重大的環境污染[2]。煤焦油加氫精制生產清潔燃料是煤焦油產業發展的確實有效出路。煤焦油加氫精制工藝涵蓋了多個復雜的反應過程,主要有加氫脫硫(HDS)、加氫脫氮(HDN)、加氫脫氧(HDO)、加氫脫金屬(HDM)、加氫裂化(HDC)等反應過程[3]。
煤焦油與渣油、頁巖油有著眾多的相似之處,目前,煤焦油的餾分加氫多采用渣油加氫催化劑進行加氫反應,但是煤焦油和渣油在N、S 含量(質量分數)上有著很大的差別。煤焦油中氮含量比硫含量多,渣油中硫含量比氮含量高,且煤焦油中硫氮含量都較渣油中硫氮含量高,尤其是氮含量,往往都在1%(質量分數)以上,所以渣油加氫催化劑應用于煤焦油加氫效果不太理想[4]。而且大量文獻資料及應用實例表明,少量氮化物就能抑制催化劑的深度加氫脫硫、加氫脫芳,還會對加氫脫氮產生自抑制現象[5];C—N、C=N 的鍵能大于C—S,煤焦油加氫脫氮的難度比加氫脫硫要高。因此,開發適用于煤焦油加氫精制的新型高效加氫脫氮催化劑,從而生產清潔燃料是非常很有必要的。本文主要對煤焦油中含氮化合物分布、典型含氮化合物HDN 反應網絡、加氫脫氮催化劑研究進展進行綜述。
1 煤焦油中含氮化合物的分布
煤焦油中化合物數量非常龐大,其中存在大量的有機含氮化合物。煤焦油中現在已鑒定出來的500 多種化合物中,含氮化合物就已經超過了120種[6],其類型可以分為:①鹽基化合物,也稱焦油鹽基,主要是雜環化合物(吡啶、喹啉及其衍生物)、芳香胺(苯胺及其衍生物),在高沸點餾分中還發現了萘胺;②中性化合物,吡咯類衍生物(吲哚、咔唑、苯并咔唑等)以及腈類化合物(苯腈、甲苯腈和萘腈等)。煤焦油中各種含氮化合物的組分含量、化學及物理性質的確定是很重要的,是煤焦油資源高效綜合利用以及利用各種化學物理方法脫除氮硫化合物而降低污染的一個非常重要的前提。尤其是在加氫脫氮催化劑研制過程中,根據其不同組分含量研究具有高原料適應性的催化劑,最終提高煤焦油的附加值和催化劑的選擇性以及壽命。煤焦油、石油、渣油中各元素分析見表1[7],煤焦油中主要含氮化合物含量見表2。
2 煤焦油中典型含氮化合物及其HDN 網絡
2.1 吡啶
吡啶在煤焦油中的質量分數約為0.03%,是比較典型的含氮化合物。吡啶的主反應網絡如圖1 所示。吡啶(PYR)加氫脫氮反應首先是由吡啶加氫生成哌啶(PIP),隨后一個C—N 鍵斷裂生成戊胺(PA),戊胺在脫去NH3生成1-戊烯(1-PENT),最后雙鍵飽和生成戊烷。此外副反應還會生成正戊基哌啶、異戊基哌啶[8]。Jan 等[9]研究了(Ni)WP/SiO2催化劑對吡啶加氫脫氮,研究中發現在T<600K 時,PYR 在WP 和NiWP 催化下PIP 的轉化率都很大,而PA、PENT 的產率則很少。T=633K 時,NiWP對PENT 選擇性比WP 高出3%。當催化劑的量增加時,產物的產率都相應增加,NiWP 和WP 相比,前者PYR 的轉化量是后者的兩倍多,而且能生成更多的PA 和PENT。在吡啶的加氫脫氮反應中,k2是整個反應的限速步驟。

表1 煤焦油、石油、渣油元素分析
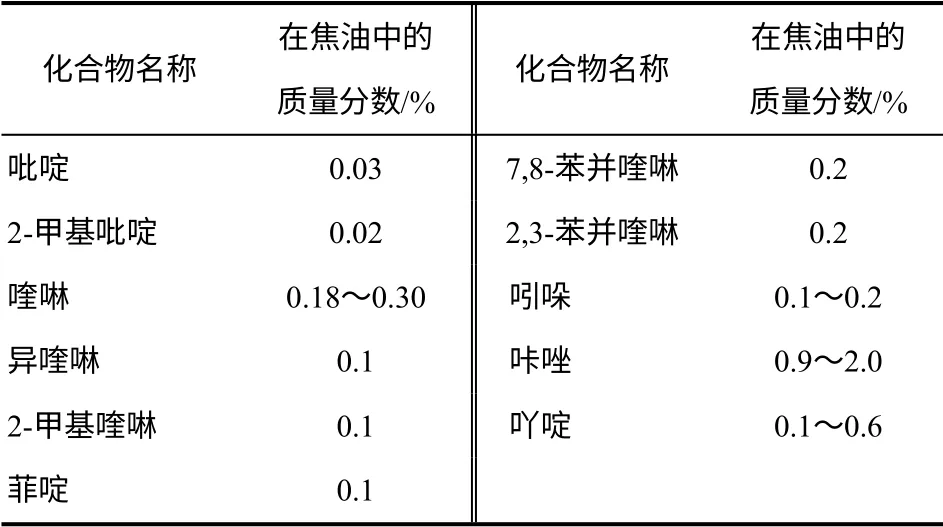
表2 煤焦油中主要含氮化合物含量(質量分數)

圖1 吡啶HDN 的主反應網絡
2.2 喹啉
喹啉(Q)及其同系物在煤焦油中的質量分數在0.4%~0.6%,其反應網絡和主要產物如圖2 所示。在喹啉HDN 反應中,THQ1 和THQ5 是主要的中間產物,說明Q 轉化THQ1 和THQ5 能夠快速地達到化學平衡。欒業志[10]研究NiW/γ-Al2O3催化劑下喹啉HDN 反應,由于氮雜環電子云密度大的緣故,在τ=0.56(LHSV)時,THQ1 的選擇性是THQ5的9 倍,反應主要以Q→THQ1 路徑進行,也可以認為THQ1 和THQ5→DHQ 是整個反應的限速步驟,在液體產物中并沒有檢測到PCHA 存在,說明脂肪烴中的C—N 鍵很容易斷裂,脫氮速率非常快。許多研究者已經發現,Q、THQ1、THQ5 的存在極大抑制了OPA 的反應,由于自抑制效應的存在,對加氫也是不利的[11]。
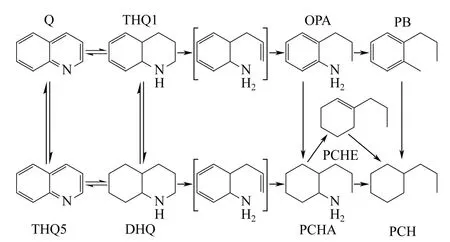
圖2 喹啉HDN 反應網絡
Infantes-Molina 等[12]研究發現,在MoP-d/r550催化劑進行HDS+(HDN)反應時,反應溫度T>623K時,添加噻吩(200μL/L)對Q 的轉化是有利的,噻吩HDS 生成的H2S 加速了PIP 的氫解,同時也加快了整個HDN 反應的總體速率。
2.3 咔唑
咔唑類化合物具有復雜的分子結構,空間位阻大,同時與堿性含氮雜環化合物存在競爭反應[13],所以在HDN 反應中較難脫除N 原子。咔唑HDN反應網絡如圖3 所示。李翠清等[14]在研究咔唑在WP 催化劑下的HDN 反應,發現咔唑HDN 反應主要包括3 個路線:k2直接脫氮生成聯苯(BP);k4先使兩個苯環加氫飽和然后脫氮,產物為環己基環己烷(DCH);k3是咔唑中其中一個苯環加氫隨后脫氮,產物為環己基苯(CHB)。3 條主要反應路線路徑中,選擇性大小順序為k4>k3>k2,這主要是因為反應物和中間產物對整個反應的自抑制效應和對活性位的強烈吸附。
2.4 吲哚
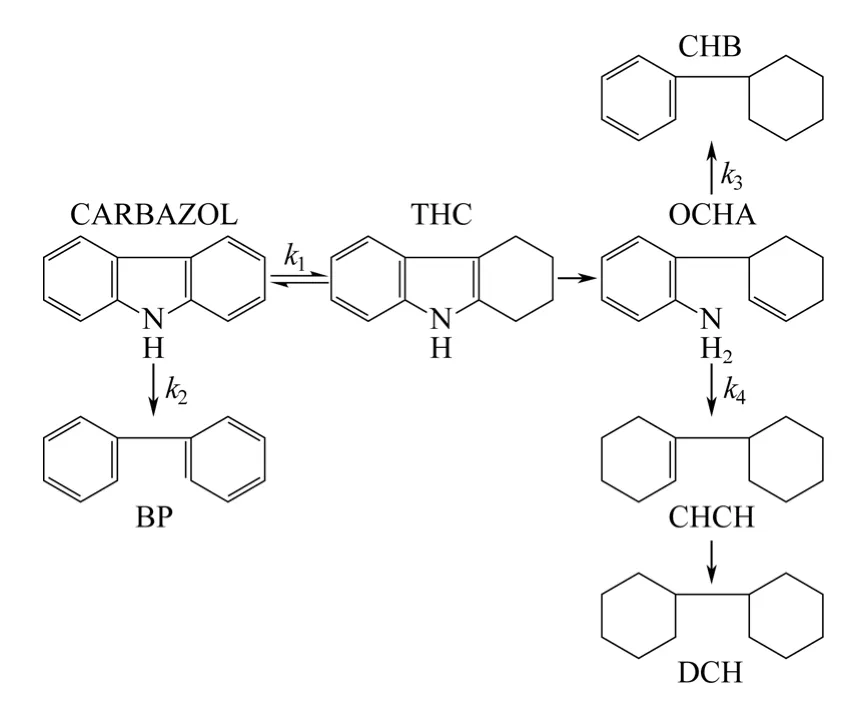
圖3 咔唑HDN 反應網絡
吲哚的加氫脫氮過程相對于芳香性六元含氮化合物要容易得多,對應的反應網絡也更加復雜,中間產物及最終產物在不同的催化劑上也不一樣。在活性組分為金屬硫化物時的反應網絡如圖4 所示[15]。一般認為吲哚加氫脫氮主要通過兩條路徑進行:首先IDN 中的吡咯環快速加氫生成HIN,隨后一條路徑為HIN 開環,脫胺,最終產物為EB 或中間產物OECHA、ECHE;另外一條路徑為HIN 飽和加氫后再開環,最終產物為ECH。Witold Piskorz等[16]研究了吲哚在Mo2C 催化劑下加氫脫氮機理,發現由于催化劑表面吸附和中間產物OEA 上π電子云密度的重新分布形成了許多的Mo—C(環)鍵,隨后H 直接進攻苯環和胺基,部分OEA 的深度脫芳形成了牢固的OEA-Mo2C-π復合體;氫的進攻選擇性及相應路徑的勢壘決定了直接脫氮(DDN)和加氫(HYD)脫氮的反應速率常數,也決定了其最終產物(EB 和ECH)的選擇性。

圖4 吲哚HDN 反應網絡
2.5 含吡啶環的雜環化合物
喹啉的衍生物在煤焦油中很多,其含量占了煤焦油含氮化合物中的大部分,其中有很多含氮化合物是同分異構體,如5,6-苯并喹啉、7,8-苯并喹啉、2,3-苯并喹啉、菲啶、吖啶等。該類化合物空間結構復雜,因此這類化合物的HDN 反應中脫除N 原子較難,菲啶及吖啶中的N 原子相對苯并喹啉更難脫除。Yeh Ji-Chang[17]的博士論文中對5,6-苯并喹啉、7,8-苯并喹啉、菲啶等含氮雜環化合物進行了HDN 的動力學研究,其中菲啶的反應網絡如圖5所示,其他化合物反應網絡可參照文獻[17]。由于空間結構的不同,菲啶的HDN 反應第一步包含了吡啶環和外部苯環的競爭加氫反應,而5,6-苯并喹啉和7,8-苯并喹啉的HDN 反應第一步只包含了對吡啶環的加氫,對于吖啶來說,吡啶環處于中間,所以在反應中首先進行的是對外部苯環的加氫反應。菲啶HDN 的中間產物八氫菲啶能夠快速地加氫生成全氫菲啶而其只能生產很小一部分的C—N鍵氫解產物(1-環己基-2-甲基苯和1-苯基-2-甲基環己烷),這與5,6-苯并喹啉和7,8-苯并喹啉為模型物HDN 的產物分布不同,這是由于八氫苯并喹啉中C—N 鍵的氫解速度要比全氫苯并喹啉及其衍生物的速率快。同樣也說明在含吡啶環的雜環化合物中,加氫脫除吡啶環位于中間的雜環化合物中的N原子要比吡啶環在外部的化合物要困難得多。

圖5 菲啶HDN 反應網絡
3 加氫脫氮催化劑
催化劑制備是一個極其復雜的過程,過去往往是憑經驗來進行催化劑的制備,隨著表面物理、表面化學、有機催化機理的深入研究以及更為先進精密的表征儀器的發明,使得各種專一性催化劑的研發變得更加容易[18]。催化劑主要由活性組分、助劑和載體三部分構成。由于煤焦油組分的復雜性,一種性能優良的煤焦油加氫催化劑要求具有適宜孔徑和活性金屬含量、分散均勻、催化活性高、選擇性好、穩定性好、耐中毒和抗積炭能力強以及對來料適應性強等特點。
3.1 加氫脫氮機理研究
煤焦油中的含硫化合物在加氫脫硫(HDS)中可以不通過飽和加氫即可脫除化合物中的S 原子,但是大部分含氮化合物必須通過加氫飽和后才能脫去N 原子。因此,芳香族含氮化合物加氫脫氮反應(HDN)可以分為兩個步驟[19]:首先通過對芳香族含氮化合物加氫使得鍵能較強的芳香族C—N,C=N 轉化為鍵能相對較弱的脂肪族C—N 鍵,再通過取代或者消除反應除去N 雜原子。
Yang 等[20]在研究活性組分為金屬硫化物的HDN 反應中發現,Ni-Mo/Al2O3催化劑在喹啉的HDN 反應中同時提供了兩類活性中心:一類是Mo上的缺硫結構的活性中心,該活性中心能夠加氫并且可能有氫解的能力;另一類是B 酸中心,B 酸是氫解和芳烴異構化的活性中心。即加氫和C—N 鍵斷裂是發生在不同的活性位上的。以金屬硫化物為活性組分HDN 反應機理的研究相對成熟,對于金屬氮、碳、磷化物的HDN 研究目前主要集中在反應活性和反應網絡上,其HDN 機理研究雖然發現部分是與金屬硫化物的機理相似,但是其更深層次的機理還需要做大量的研究。
含氮化合物中的胺類的加氫脫氮(HDN)反應機理最早是用Hoffman 消除與SN親核取代反應來對反應過程進行解釋的[21]。HDN 機理認為首先經過飽和加氫的分子接受B 酸中心提供的H+形成相應的氮正離子,然后與B-反應(消除或者取代反應),對應著生成烯烴、硫醇和NH3。Zhao 等[22]和Portefaix等[23]在C—N 鍵氫解上又提出了新的機理,認為該鍵的氫解取決于C—N 鍵上C 的類型,并不是α-C、β-C 上H 的類型與數量。
隨著金屬有機化學的發展,1983 年,Laine[24]提出了一個從金屬有機化學角度解釋C—N 斷裂機理,認為對加氫飽和后的含氮化合物起決定作用的是金屬原子(離子)而并不是Nelson 等[21]提出的酸性位。該機理認為C—N 鍵可以通過兩種方式斷裂:一是吡啶生成環狀烷基金屬物種;二是可以絡合形成金屬卡賓物種。隨后經H2S 進攻不同的兩種中間物種,促使C—N 鍵斷裂。
3.2 活性組分
傳統加氫活性組分包括貴金屬和非貴金屬硫化物,通常傳統加氫活性組分包括貴金屬和非貴金屬硫化物,它們包括VIB 族的鉬(Mo)、鎢(W)以及Ⅷ族的鈷(Co)、鎳(Ni)、鐵(Fe)、鈀(Pd)、鉑(Pt)。該類活性組分中的金屬元素d 軌道呈未充滿狀態,在晶體結構上具有體心或面心、立方晶格或六方晶格,因此,從電子排布和空間結構上看這類金屬都具備作為加氫脫氮(HDN)催化劑活性組分的條件[25],然而單一的活性組分催化性能不高,需在多元活性組分下才能夠具備較高的催化活性,因而目前商業加氫精制催化劑的活性組分基本上都是由VIB 族金屬與Ⅷ族金屬組合的二元(三元)活性組分所構成,再根據原料的差異性對催化劑配級使用。在加氫脫氮反應中,不同金屬硫化物的活性為:純硫化物加氫脫氮活性大小依次為Mo>W>Ni>Co,最佳加氫脫氮組合活性順序為Ni-W≥Ni-Mo>Co-Mo>Co-W[7]。
貴金屬活性高,但是負載大量的貴金屬催化劑成本較高,人們一直在尋找一種夠保持貴金屬的高活性,同時又能夠減少貴金屬用量的一種方法。在這方面劉寬宗等[18]提到了一種納米催化劑制備技術來減少貴金屬的使用量,如圖6 所示,該方法能夠有效地減少貴金屬的移動,使催化劑保持在微粒狀態,在高溫下不易燒結,避免固態變換,金屬-載體間的作用力適中能夠在保持催化劑活性的前提下大量降低貴金屬的使用量。
除了金屬硫化物和貴金屬外,在反應中表現出高活性并且被譽為“準鉑催化劑”的金屬碳化物和氮化物受到越來越多的研究者關注和研究[26]。MoC2對喹啉(Q)HDN 活性明顯高于Mo 的硫化物及氧化物對喹啉的HDN 活性[27]; 在考察脫氮性能中,Mo2N/Al2O3的催化活性均高于MoS2/Al2O3和Co-Mo/Al2O3催化劑,Mo2N 對C—N 鍵具有更強的選擇性[28]。過渡金屬磷化物與上述氮化物和碳化物具有相似的物理性質,具有優越的加氫脫硫和加氫脫氮性能[29]。Ni2P 和MoP 是兩種較為理想的加氫脫氮金屬磷化物催化劑。雖然過渡金屬氮化物、碳化物、磷化物具有較高的催化活性,但由于其制備條件復雜苛刻、副反應多、成本高等原因,至今仍未見工業化報道。過渡金屬氮化物、碳化物、磷化物仍然是近年來的研究熱點,若能夠有效攻克該類催化劑的各種瓶頸,將會給整個催化加氫行業帶來革命性的變革。

圖6 一種減少貴金屬用量的納米催化劑制備技術
3.3 載體
載體是是催化劑中主催化劑和助催化劑的分散劑、黏合劑和支撐體,同時催化劑載體還發揮著穩定化、傳熱、稀釋、助催化劑等作用。加氫脫氮反應受擴散控制的影響,所以高效的HDN 催化劑載體應該具有適宜的比表面積、孔容以及孔徑,高比表面積和大孔徑相比之下,大孔徑更為重要,高比表面積并不能在任何條件下都能提高催化劑的活性。在加氫脫氮反應中,催化劑載體主要孔徑分布在100~300nm 時,脫氮更為有效。選擇一個合適的載體,對活性金屬有適當結合力是非常關鍵的。
3.3.1 單一載體
常見的單一載體有三氧化二鋁、二氧化硅、二氧化鋯、二氧化鈦等。氧化鋁是目前工業催化劑最常用的載體之一,目前已發現有9 種不同的晶態[30],尤以γ-Al2O3活性最好,所以也最為常用。氧化鋁具有可調的孔結構、較大的比表面積、良好的吸附性能、較好的力學性能、表面具酸性和穩定性好等優點,但是由于該載體容易與催化劑活性組分形成較強的相互作用,抑制了催化劑的活性,且該載體有較多的L 酸,缺少B 酸,通常要經過改性后才能達到優越的催化性能。王永剛等[31]研究了γ-Al2O3負載NiW 催化劑對低溫煤焦油的加氫性能,研究發現活性組分能夠良好地分散在載體表面,Ni/W=0.38 時其催化加氫活性最好,對煤焦油的加氫脫硫(HDS)和加氫脫氮(HDN)效果也是最好的,研究表面NiW/γ-Al2O3催化劑能夠在煤焦油加氫改質、變廢為寶、提高產品價值和煤焦油產業綠色發展方面有著很大的應用前景。
3.3.2 復合氧化物載體
復合氧化物載體是兩種或者兩種以上的金屬氧化物或者非金屬氧化物組成的復雜多元氧化物,各氧化物的含量能夠影響催化劑的加氫活性。復合氧化物載體可以分為天然和人工合成兩種,可作為載體的天然復合氧化物主要有凹凸棒、膨潤土、硅藻土等,常見的人工合成復合氧化物有TiO2-Al2O3、
TiO2-ZrO2、SiO2-Al2O3、SiO2-TiO2-ZrO2、SiO2-TiO2-Al2O3等。復合氧化物載體的優勢在于能保持單一載體具有的優勢,同時克服單一載體的劣勢。Al2O3載體會與活性組分形成較強的相互作用,且缺少B酸,而TiO2載體的比表面積小,熱穩定性差,但是當使用TiO2-Al2O3作為載體時,既保持了Al2O3的高比表面積和高熱穩定性等優點,TiO2的引入能夠減弱Al2O3與金屬硫化物和金屬磷化物(活性組分)之間的相互作用,提高前體的還原能力,形成更多的活性物,TiO2能夠作為Ni-P 催化劑的電子助劑,能夠提供較多B 酸中心,并且能夠有效地促進C—N 的斷裂及加氫的活性;不同的Ti/Al 比對喹啉的HDN 活性影響很大, 當Ti/Al=1/8 時,Ni2P/TiO2-Al2O3對喹啉的HDN 活性最佳,在這里TiO2不僅能夠作為載體,還具有助催化劑作用[32]。
對于活性組分為金屬硫化物的催化劑來說,TiO2的引入對前體的硫化和還原是有利的。石芳等[33]研究了以W-Mo-Ni/SiO2-TiO2-Al2O3催化劑對高氮餾分油的加氫精制,SiO2和TiO2引入并沒有降低載體的熱穩定性和載體的比表面積以及孔容;促進了WO3和MoO3相還原硫化為具有催化活性的WS2和MoS2相;三元載體中各氧化物能夠調變載體的酸性,減弱一些含氮化合物對HDS 的抑制和金屬-載體的相互作用,從而提高餾分油的HDN 和HDS 活性。研究結果表明,W-Mo-Ni/SiO2-TiO2-Al2O3具有較高的HDN、HDS、烯烴加氫飽和活性,脫氮率為97%,脫硫率為98%,飽和率為98%。
3.3.3 碳載體
碳載體具有巨大的比表面積,孔結構分布寬,擁有良好的熱、化學穩定性,作為一種新型的催化劑載體,其對活性組分和助劑表現出優越的分散性能,碳載體來源廣泛,價格低廉。目前常見的碳載體有活性炭(AC)、介孔碳材料(mC)和碳納米管(CNTs)。Shi 等[34]利用油砂石油焦制取了活性炭,研究了Ni-Mo/AC 對重質減壓瓦斯油(HVGO)加氫處理,利用兩種不同的原料制備兩種活性炭(FAC,DAC)與兩種商品活性炭及氧化鋁作為載體,研究表明自制活性炭載體的加氫催化活性較其他載體相比具有更高的活性,康氏殘炭值(CCR)較低、油品的HDS 及HDN 轉化率均比其他種類催化劑好。Ni-Mo/DAC 催化劑較Ni-Mo/FAC 催化劑表現出更加高的活性。EXD 圖像表明活化后的活性組分粒子均勻地分散在活性炭的孔道內,形成眾多的活性位,使該催化劑具有高的HDS 和HDN 活性。Prabhu 等[35]以NiMo/mC 為催化劑對輕油的加氫脫氮和加氫脫硫進行了研究,發現由于介孔碳載體的高比表面積和與活性組分(NiMoS)的弱相互作用,使NiMo/mC 的HDN 和HDS 活性高于傳統的NiMo/γ-Al2O3。Dong 等[36]在以Co-Mo-S/CNTs 為催化劑對吡咯的HDN 反應中發現,與活性炭載體形成的活性種數量相比,以碳納米管為載體的表面能形成大量的Mo 活性種(Mo4+),另外該功能催化劑能夠在特定的壓力和不同的溫度下(室溫至673K)內可逆地吸附大量的氫,這一獨特的特征使得H 能夠在催化劑表面形成大量的H 活性種,這些特點使得該催化劑具有較高的催化活性。Kong 等[37]以Pt/CNTs對甲苯和四氫化萘的加氫脫芳(HDA)中得到了同樣的結果。
3.3.4 介孔材料和分子篩載體
介孔材料和分子篩載體由于具有巨大的表面積,孔結構規整,組成、性質、孔徑大小、酸性可調,有良好的熱穩定性和水熱穩定性,并且能夠使得活性組分高度分散,增加活性相的數量。常見的分子篩載體有NaY、MCM-41、SAPO-5、USY、ZSM-5、SBA-15 等,在石油煉制中以Y 型分子篩、SBA-15等分子篩最為常用。
魏強等[38]在Y 型分子篩中引入P 合成PY 分子篩,P 的引入并沒有破壞Y 的結構,PY 分子篩保持了完整的晶型,使分子篩的強B 酸和總酸量下降,最終使該新分子篩具有較大的孔徑、孔容和合理的酸分布,使催化劑加氫精制活性得到明顯提高。Yin等[39]研究表明,在含微米和納米尺寸的Y 型分子篩的NiMo/Al2O3催化劑對柴油的加氫精制活性明顯提高,表現出良好的尺寸效應,其HDN、HDS 效果均優于微米分子篩。SBA-15 具備作為催化劑載體具有優異的結構特性,但是其缺少表面酸和金屬-載體相互作用弱,限制了其更廣泛的應用。以Zr改性的SBA-15 具有合適的表面酸和金屬-載體相互作用,Zr-SBA-15 可以作為一種有效的加氫催化劑載 體 。 NiMo/Zr-SBA-15 、 NiMo/SBA-15 、NiMo/γ-Al2O3三種催化劑對重瓦斯油的HDN 和HDS 活性考察結果表明,Zr-SBA-15 負載的催化劑的活性要高于其他兩種[40]。
3.4 助劑
助劑在催化劑中的含量只占很少一部分,但是卻能夠極大地提高催化劑的催化活性。助劑能夠在調變金屬-載體相互作用、催化劑酸量及酸分布、孔徑及比表面積等[18]產生很大影響,從而增加比表面積、選擇性、活性組分的分散度、活性相的數量,改變活性相的結構,具備適宜的酸度和孔徑,最終使得催化活性顯著提高。目前可利用于加氫精制的助劑是多種多樣的,大致可以分為兩類:非金屬助劑P、F、B 等;金屬助劑Co、Ni、Ti、Fe、V、Ce、Mn、Cr、Pt、Pd、Ir、Os、Rh、Re 等[3]。上述助劑都能夠在加氫脫氮反應中提高催化劑的性能。
P 及金屬磷化物的引入能使金屬-載體的相互作用減弱,活性組分的分散度及活性位數量增加。P 元素引入還能夠與Al2O3載體形成AlPO4相,從而使活性組分硫化更容易,形成更多活性位,使得HDN 活性增加[41]。P 的引入能夠對NiMo/γ-Al2O3催化劑活性相結構產生很大的影響,能夠降低四面體配位的Mo 物種數量,增加八面體配位(高活性)的Mo 數量,適當的磷助劑能夠增加邊角位有效的Mo 和活性位數量[42]。Liu 等[11]研究了Ni2P 與MoS2在喹啉HDN 反應的協同效應,發現Ni2P 的引入提高了MoS2活性位的數量,使得該催化劑對喹啉的速率及脫氮活性顯著提高,Ni2P 表現出優異的助催化劑活性。
硼能夠調變催化劑的表面酸性,隨著助劑硼的含量增加,B 酸增多而L 酸減少,使得活性組分的結晶度增加,能夠增加催化劑的HDN 活性,當B的質量分數為5%時,對減壓蠟油的脫氮率為77%,比未引入硼時脫氮率增加15%[43]。Yao 等[44]以NiMo/Al2O3催化劑添加F、B 雙助劑對循環輕油加氫研究,B 可能會取代載體中部分Al3+形成B3+,也可以形成B—OH 或A1—O—B—O—A1 就能夠改變載體的酸性,F、B 的引入能夠調節載體的酸性和Ni、Mo 的分散度,直接影響催化劑的HDN、HDS、HDA 活性,添加不同量的F、B 能夠改變催化劑的加氫活性,不同催化劑的HDN 活性大小順序為:NiMo/F,B-Al(3.5)>NiMo/F,B-Al(5.0)≈NiMo/F,B-Al(7.0)。
傳統Ni、Co 在加氫精制領域應用最廣泛,Ni在加氫脫氮反應中作用顯著。在WP/SiO2催化劑中添加Ni 助劑,在對吡啶的HDN 反應中,Ni 的引入能夠促進吡啶加氫和C—N 斷裂,使得吡啶HDN轉化率增加,同時使生成戊烷的選擇性增加[9]。劉維橋等[45]在WNx催化劑引入Ni,結果使催化劑前體還原氮化更容易,而且使W 活性位更加穩定存在。V 作為助劑也能夠使含氮化合物HDN 反應的轉化率顯著提高,當V 負載量為1%時,催化劑的HDN 活性提高最為顯著[14]。Sun 等[46]在Ni2P 引入CeO2能夠增加Ni2P 的比表面積和CO 吸附,使其加氫和C—N 鍵斷裂活性顯著增加,CeO2能夠提高喹啉生成丙基環己烷的選擇性。
4 展 望
煤焦油加氫脫氮(HDN)催化劑應該以高活性、高穩定性及高原料適應性的方向進行研究,應對加氫脫氮催化劑從以下幾個方面進行進一步研究。
(1)載體 對載體進行改性,獲得適宜的酸性、孔徑、孔容,使得活性組分能夠高度分散在載體表面。
(2)活性組分 金屬硫化物目前仍然是加氫精制的主要活性組分,其加氫脫氮活性的提高可以通過不同的制備方法和配級來實現。金屬碳、氮、磷化物表現出優異的HDN 活性,要實現工業化應用,其制備方法、穩定性等方面需要進一步研究。
(3)助劑 除了傳統助劑以外,要把加氫脫氮助劑關注點放在貴金屬、稀土金屬以及各種過渡金屬氮、碳、磷化物上,這一類物質作為助劑能夠使催化劑的活性顯著增加。
煤焦油加氫精制制取低N、S 的燃料油品是提高煤焦油附加值的一種有效方法,對煤焦油中含氮化合物的HDN 機理、反應網絡、動力學等方面進行深入研究,研制高效HDN 催化劑,不僅能夠得到低氮油品,同時能夠減輕深度脫硫、深度脫芳的抑制,有利于煤焦油開發成為一種清潔能源,降低其對環境的污染,使煤焦油產業朝著綠色化方向發展。
[1] 張軍民,劉弓. 低溫煤焦油的綜合利用[J]. 煤炭轉化,2010,33(3):92-96.
[2] 黃新龍,孫殿成,王洪彬,等. 高溫煤焦油餾分油加氫改質生產清潔燃料研究[J]. 煤炭轉化,2013,36(1):79-83.
[3] 李矗,王安杰,魯墨弘,等. 加氫脫氮反應與加氫脫氮催化劑的研究進展[J]. 化工進展,2003,22(6):583-588.
[4] 陳松,許杰,方向晨. 煤焦油聯合加氫裂化處理工藝及其專用催化劑[J]. 現代化工,2009,29(3):64-68.
[5] 鄭云弟,蔣彩蘭,康宏敏,等. 加氫脫氮催化劑載體的研究[J]. 工業催化,2010,18(6):1-7.
[6] 肖瑞華. 煤焦油化工學[M]. 北京:冶金工業出版社,2009:401-418.
[7] 馬寶岐,任沛建,楊占彪,等. 煤焦油制燃料油品[M]. 北京:化學工業出版社,2010:101-103.
[8] Wang Huamin,Prins Roel. On the formation of pentylpiperidine in the hydrodenitrogenation of pyridine[J].Catal.Lett.,2008,126:1-9.
[9] Jan Kopyscinski,Jinsoon Choi,Josephine M Hill.Comprehensive kinetic study for pyridine hydrodenitrogenation on (Ni)WP/SiO2catalysts[J].Applied Catalysis A:General,2012,445-446:50-60.
[10] 欒業志. 撫順頁巖油組分分析和喹啉在NiW/γ-Al2O3催化劑上的加氫脫氮研究[D]. 大連:大連理工大學,2009.
[11] Liu Lihua,Liu Shugun,Chai Yongming,et al. Synergetic effect between Ni2P/γ-Al2O3and MoS2/γ-Al2O3catalysts on their performance in hydrodenitrogenation of quinoline[J]. J. Fuel Chem.Technol.,2013,41(6):698-702.
[12] Infantes-Molina A,Moreno-Leon C,Pawelec B,et al.Simultaneoushydrodesulfurization and hydrodenitrogenation on MoP/SiO2catalysts:Effect of catalyst preparation method[J]. Applied Catalysis B:Environmental,2012,113-114:87-99.
[13] 路蒙蒙,孫守華,丁保宏,等. 加氫脫氮反應研究進展[J]. 化工科技,2011,19(1):65-71.
[14] 李翠清,孫桂大,李鳳艷,等. 釩摻雜的磷化鎢催化劑咔唑加氫脫氮性能[J]. 石油化工高等學校學報,2009,22(3):24-27.
[15] 張奇. 吲哚在NiW/γ-Al2O3催化劑上加氫脫氮的研究[D]. 大連:大連理工大學,2010.
[16] Witold Piskorz ,Grzegorz Adamski,Andrrzej Kotarba,et al.Hydrodenitrogenation of indole over Mo2C catalyst:Insights into mechanistic events through DFT modeling[J].Catalysis Today,2007,119 :39-43.
[17] Yeh Ji-Chang. Hydrodenitrogenation studies of condensed carbocyclic-N-heterocyclic compounds and of an SRC-II distillate[D].Salt Lake City:University of Utah,1989.
[18] 劉寬宗,張磊,江健,等. 煤焦油加氫精制和加氫裂化催化劑的研究進展[J]. 化工進展,2012,31(12):2672-2677.
[19] Wahyudiono,Yui Matsunaga,Siti Machmudah,et al.Supercritical water as a reaction medium for nitrogen-containing heterocycles[J]. J.Chem.Chem.Eng.,2012,10:897-910.
[20] Yang S H,Saterfield C N.Catalytic hydrodenitrigenation of quinoline in a trickle-bed reaction effect of hydrogen sulfide[J].Ind.Eng.Chem.Proc.Dev.,1984 ,23(1):20-25.
[21] Nelson N,Levy R B.The organic chemistry of hydrodenitrogenation[J].Journal of Catalysis,1979,58(3):485-488.
[22] Zhao Y , Prins R. Mechanisms of the hydrodenitrogenation of alkylamines with secondary and tertiary a-carbon atoms on sulfided NiMol/Al2O3[J].J.Catal.,2004,222(2):532-544.
[23] Portefaix J L,Cattenot M,Guerriche M,et al. Mechanism of carbon-nitrogen bond cleavage during amylamine hdrodenitrogenation over a sulphided NiMol/A12O3catalyst[J].Catal.Lett.,1991,9(1-2):127-132.
[24] Laine R M.Comments on the mechanisms of heterogeneous catalysis of the hydrodenitrogenation reaction[J]. Catalysis Reviews:Science and Engineering,1983,25(3):469-474.
[25] 宣揚. 催化裂化原料油加氫脫氮催化劑研究[D]. 青島:中國石油大學(華東),2011.
[26] 張國輝,劉紅光,楊建國,等. 清潔柴油加氫精制催化劑的技術進展[J]. 精細石油化工,2011,28(4):77-82.
[27] 羅運強,靳廣洲. 碳化鉬催化劑的喹啉加氫脫氮反應性能[J]. 工業催化,2009,17(11):16-21.
[28] 張新波,張雅娟. 過渡金屬氮化物催化性能研究進展[J]. 工業催化,2011,19(1):11-15.
[29] 顧永和,王博,崔操,等. 反應條件對MoP/TiO2-SiO2-A12O3催化劑加氫脫氮性能的影響[J]. 化學與黏合,2013,35(6):46-49.
[30] 盧一鳴. 不同晶型氧化鋁與制備工藝對YAG:Ce3+光學性能的影響的研究[D]. 上海:上海師范大學,2012.
[31] 王永剛,張海永,張培忠,等.NiW/γ-Al2O3催化劑的低溫煤焦油加氫性能研究[J]. 燃料化學學報,2012,40(12):1492-1497.
[32] Yan J S,Wang H Y,He F W.Preparation of TiO2-Al2O3support and hydrodenitrogenation over Ni2P/TiO2-Al2O3catalyst[J]. Advanced Materials Research,2014,881:245-250.
[33] 石芳,楊建國,劉紅光,等. 高氮餾分油加氫精制催化劑的研制[J].精細石油化工,2011,28(6):44-48.
[34] Shi Y,Chen J,Chen J,et al. Preparation and evaluation of hydrotreating catalysts based on activated carbon derived from oil sand petroleum coke[J].Applied Catalysis A:General,2012,441:99-107.
[35] Prabhu N , Dalai A K , Adjaye J. Hydrodesulphurization and hydrodenitrogenation of light gas oil using NiMo catalyst supported on functionalized mesoporous carbon[J]. Applied Catalysis A :General,2011,401(1):1-11.
[36] Dong Kunming,Ma Xiaoming,Zhang Hongbin,et al. Novel MWCNT-support for Co-Mo sulfide catalyst in HDS of thiophene and HDN of pyrrole[J].Journal of Natural Gas Chemistry,2006,15(1):28-37.
[37] Kong H,Zhou M,Lin G D,et al. Pt catalyst supported on multi-walled carbon nanotubes for hydrogenation-dearomatization of toluene and tetralin[J].Catalysis Letters,2010,135(1-2):83-90.
[38] 魏強,周亞松,陶秀娟,等.PY 分子篩為載體催化劑的加氫精制性能[J]. 石油學報,2012,28(1):15-20.
[39] Yin H,Zhou T,Liu Y,et al. NiMo/Al2O3catalyst containing nano-sized zeolite Y for deep hydrodesulfurization and hydrodenitrogenation of diesel[J].Journal of Natural Gas Chemistry,2011,20(4):441-448.
[40] Biswas P , Narayanasarma P , Kotikalapudi C M , et al.Characterization and activity of ZrO2doped SBA-15 supported NiMo catalysts for HDS and HDN of bitumen derived heavy gas oil[J].Industrial & Engineering Chemistry Research,2011,50(13):7882-7895.
[41] Ho T C,Pan W H,Jacobson A J,et al. Hydroprocessing on chromium-containing metal sulfide catalysts[J].Applied Catalysis A:General,2012,421:38-47.
[42] 周同娜,尹海亮,柳云騏,等. 磷含量對NiMo/γ-Al2O3催化劑活性相結構的影響[J]. 燃料化學學報,2010,38(1):69-74.
[43] 樊慧麗,段愛軍,趙震,等. 催化裂化汽油加氫精制催化劑的研究進展[J]. 工業催化,2011,19(11):8-13.
[44] Yao Songdong,Zheng Ying,Ng S,et al.Co-promotion of fluorine and boron on NiMoAl2O3for hydrotreating light cycle oil[J]. Catal. Sci.Technol.,2012,2:1925-1932.
[45] 劉維橋,周虎,雷衛寧,等. 助劑對WNx/TiO2-γ-Al2O3催化劑物化性質及加氫脫硫性能影響[J]. 化學學報,011,69(14):1622-1626.
[46] Sun Z , Li X , Wang A , et al. The effect of CeO2on the hydrodenitrogenation performance of bulk Ni2P[J]. Topicsin Catalysis,2012,55(14-15):1010-1021.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
