Pt/USY 催化劑上FCC 柴油加氫改質反應性能
2015-07-25 09:09:48遲克彬趙震田志堅閻立軍馬懷軍羅琛劉堅
化工進展 2015年5期
關鍵詞:催化劑
遲克彬,趙震,田志堅,閻立軍,馬懷軍,羅琛,劉堅
(1 中國石油大學(北京)理學院,北京 102249;2 中國科學院大連化學物理研究所,遼寧 大連 116023;3 中國石油石油化工研究院,北京 100195)
隨著環保法規的日益嚴格,我國制定了等同于歐Ⅳ、歐Ⅴ燃油標準的國Ⅳ、國Ⅴ標準,明確要求2014 年底車用柴油全部達到國Ⅳ標準,2017 年底車用柴油全部達到國Ⅴ標準。而目前的現狀是,我國柴油池中FCC 柴油所占比例約為30%,其密度大,硫含量和芳烴含量較高,十六烷值較低[1-2],使得國內各煉化企業在柴油質量升級過程中面臨嚴峻的挑戰。要達到國Ⅴ柴油質量標準,除了更嚴格的脫硫要求以外,不同標號柴油的十六烷值至少還要增加2 個單位,目前加工中間基原油和環烷基原油的煉廠在生產十六烷值為49 的柴油時已經非常困難,即使加工石蠟基原油的企業,因為其催化裂化裝置大都采用MIP 工藝,柴油的十六烷值很低,也不得不通過加氫改質、混兌、摻煉、切割等各種損失柴油收率的手段來滿足柴油十六烷值出廠要求。
柴油十六烷值、密度與烴類族組成密切相關[3],相同碳數不同族組成烴類的十六烷值按以下順序遞減:正構烷烴>烯烴>異構烷烴>環烷烴>芳烴,其中碳鏈分支越多十六烷值越低,芳環越多十六烷值也越低,但芳環帶長側鏈可提高其十六烷值;同一烴類的十六烷值隨著碳原子數的增加而增加[4-5]。目前,提高FCC 柴油質量的技術主要有加氫飽和(ASAT)、加氫裂化(HC)以及芳烴和環烷烴的選擇性開環(SRO)[6-10]。加氫飽和技術通過將FCC柴油中的芳烴飽和為環烷烴提高柴油的十六烷值,但其增幅有限,且密度降低幅度較小;加氫裂化技術主要通過將FCC 柴油中的稠環芳烴或環烷烴非選擇性裂化為單環芳烴、環烷烴或鏈烷烴類,達到提高柴油十六烷值和降低柴油密度的目的,但是以犧牲柴油收率為代價;而芳烴和環烷烴的選擇性開環技術則主要是先將稠環芳烴飽和為環烷烴,然后再將環烷烴選擇性開環,得到帶長側鏈的環烷烴(反應歷程如圖1[11]所示),從而在不減少柴油收率的前提下大幅度提高柴油的十六烷值,同時降低了柴油密度。與加氫飽和或加氫裂化技術相比,芳烴選擇性開環技術在大幅度提高柴油十六烷值的同時,兼顧了較高的柴油收率。因此,芳烴選擇性開環是FCC 柴油加氫改質反應的最佳技術[12]。本文以預精制FCC 柴油為原料,采用芳烴選擇性開環反應,生產滿足國Ⅴ標準的柴油餾分,詳細研究了Pt/USY催化劑的酸性對芳烴選擇性開環反應的影響規律,并初步探討了十六烷值與烴類族組成的關系。
1 實驗部分
1.1 催化劑制備
H-USY 分子篩由蘭州石化公司提供,其SiO2/Al2O3摩爾比為5.8。以一定濃度的KNO3水溶液初濕浸漬USY 分子篩,得到K 質量分數分別為1.0%和2.0%的兩種分子篩,標記為1.0K-USY 和2.0K-USY,然后將USY、1.0K-USY 和2.0K-USY這3 種分子篩原粉分別按相同比例與擬薄水鋁石及黏合劑混合均勻、攪拌、擠條成型,在120℃烘干,在550℃焙燒4h,粉碎得到顆粒大小為1~2mm 的催化劑載體。用一定濃度的Pt(NH3)4Cl2溶液等容浸漬,控制鉑的負載量為0.5%(質量分數)。在120℃干燥24h、空氣氣氛下500~600℃焙燒4h 后分別制得0.5%Pt/USY、0.5%Pt/1.0K-USY 和0.5%Pt/2.0K- USY(質量分數)催化劑,標記為SRO-1、SRO-2和SRO-3。
1.2 催化劑表征
(1)X 射線衍射譜表征(XRD) 采用日本理學D/max-rb 型X 射線粉末衍射儀對實驗樣品進行物相表征。測定條件:Cu 靶,Kα射線(λ=1.5406?),Ni 濾波,工作電壓為40kV,工作電流40mA,掃描范圍 2θ 為5°~70°,掃描速度5°/min。
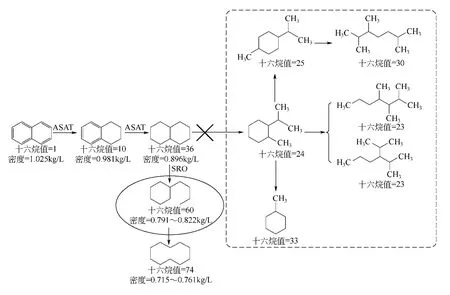
圖1 芳烴選擇性開環反應路徑[11]
(2)吡啶-紅外光譜表征(Py-IR) 樣品的Pyridine-IR 表征在美國Bruker EQUINOX55 紅外光譜儀上進行。將 10mg 左右的樣品粉末壓成10mg/cm2自支撐樣品片,固定在紅外池中,400℃溫度下抽真空(1×10-2Pa)凈化處理樣品2h,冷卻至室溫,掃描背底。吸附吡啶30min 后,程序升溫到150℃,300℃進行脫附2h,降至室溫,記錄1700~1400cm-1波數區域的紅外光譜。以1450cm-1處的峰表征L 酸,以1540cm-1處的峰表征B 酸。
(3)NH3-TPD 表征 NH3-TPD 表征在美國康塔公司生產的AUTOSORB-1 C/TCD-MS 全自動吸附儀上完成,稱取100~200mg 樣品裝入U 形石英反應管內,以 10℃/min 升溫到 550℃,通入40mL/min 的 He 氣預處理1h,冷卻至100℃吸附NH3(40mL/min)至飽和。切換到He 氣(40mL/min),吹至40℃,再程序升溫(10℃/min)至600℃下脫附,用TCD 檢測器檢測記錄信號。
(4)N2等溫吸附-脫附 采用美國Micromeritics 公司ASAP 2405M 型物理吸附儀對樣品進行N2等溫吸附-脫附分析,N2作為吸附劑,30~40mg 樣品首先在300℃、1.33×10-3Pa 條件下處理15h,然后在液氨溫度(-196℃)下進行測試。
1.3 催化劑性能評價
加氫改質評價試驗前,選用已商業化的柴油加氫精制催化劑對FCC 柴油進行預處理,得到硫含量3.93μg/g、氮含量小于0.5μg/g 的加氫預精制FCC柴油。對于貴金屬/分子篩型催化劑,FCC 柴油中硫、氮含量的高低會顯著影響貴金屬催化劑的活性和開環選擇性。高含量硫易使催化劑的貴金屬活性中心中毒,影響其加氫性能;高含量氮易使催化劑的酸性中心中毒,影響催化劑的選擇性開環性能[13]。因此,FCC 柴油在進行加氫改質反應前必須脫除原料油中的硫、氮雜質,以滿足貴金屬催化劑的評價試驗要求。
采用固定床連續加氫反應試驗裝置研究SRO-1、SRO-2 和SRO-3 催化劑上加氫預精制FCC柴油的加氫改質反應性能。催化劑的裝填量為10mL,催化劑床層兩端填充1~2mm 的惰性瓷球。催化劑評價前,在氫氣流速15mL/min、溫度350℃條件下原位還原4h。反應條件:溫度240~300℃,H2/oil(體積比)=500~1000,液時空速(LHSV)= 2.0~4.0h-1,H2壓力4.0~8.0MPa,進料4h 待反應穩定后開始取樣。液體產物經冷卻分離,在Varian CP-3800 GC 和Agilent 5973 MS 聯用儀上,采用ASTM D2425 方法分析柴油餾分中烴類族組成;在Agilent 7890 A 氣相色譜儀上,采用ASTM D2887方法分析液體產物的餾程;采用GB/T 1884—2000方法分析柴油餾分的密度;在小型柴油機上采用ASTM D7170 方法測試柴油餾分的十六烷值。
芳烴的飽和率和開環率按式(1)和式(2) 計算。

2 結果與討論
2.1 催化劑的結構和酸性
USY 分子篩和SRO-1、SRO-2、SRO-3 催化劑的XRD 譜圖見圖2。從圖2 可知,USY 分子篩經過不同濃度的KNO3溶液改性后,即使K 質量分數高達2%,仍然沒有出現新的晶相,保持了USY 完整的晶型,結晶度較高。負載金屬Pt 制備成催化劑后,也沒有出現新的晶相,說明金屬Pt 和K 在分子篩載體表面均呈高分散狀態,沒有出現聚集現象。
表1 為SRO-1、SRO-2、SRO-3 催化劑的孔道結構數據。盡管隨著K 含量增加,催化劑的孔容積、比表面積和平均孔徑略微減小,但是3 種催化劑的孔容積、比表面積和平均孔徑仍然非常接近,這進一步證明金屬K 高分散在分子篩載體中,即使K 質量分數高達2%,仍沒有堵塞分子篩孔道。
雙功能催化劑的酸性強弱和酸中心數量對芳烴選擇性開環反應產物的分布有較大的影響[14]。圖3是SRO-1、SRO-2、SRO-3 催化劑的NH3-TPD 譜圖。由圖3 可知,SRO-1、SRO-2、SRO-3 催化劑均出現3 個脫附峰:150~250℃間的脫附峰對應弱酸中心,250~350℃間的脫附峰對應中強酸中心,350~450℃間的脫附峰對應強酸中心。從峰面積可以看出,3 種催化劑的低溫脫附峰面積均比中高溫脫附峰面積大,表明SRO-1、SRO-2、SRO-3 這3 種催化劑均以弱酸位為主;隨著K 擔載量的增加,強酸位數量逐漸減少,弱酸位數量幾乎不變;SRO-3 催化劑的酸性位幾乎全部為弱酸位。上述NH3-TPD表征結果,證明K 改性是調節USY 分子篩酸強度非常有效的方法。
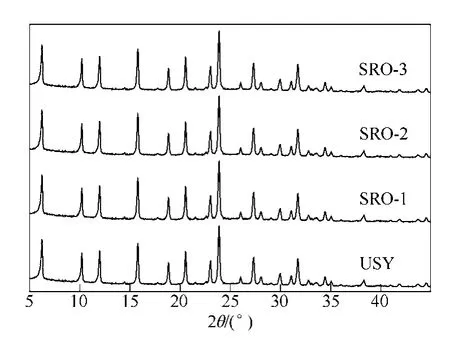
圖2 分子篩和催化劑的XRD 譜圖

表1 催化劑的物化性質
吡啶吸附紅外光譜測量的SRO-1、SRO-2、SRO-3 催化劑的B 酸和L 酸數據見表2。由表2 數據可知,SRO-1、SRO-2、SRO-3 催化劑同時含有B酸和L 酸;隨K 擔載量的增加,B 酸和L 酸數量均明顯減少。一般認為,弱酸位上吸附的吡啶在200 ℃以下進行脫附,而強酸位上吸附的吡啶在350℃高溫以上進行脫附。隨K 擔載量的增加,催化劑的強酸位數量發生顯著下降,而弱酸位的數量僅有少量的減少。催化劑Py-FT-IR 表征的結果與NH3-TPD結果具有一致性。

圖3 Pt/USY 催化劑的NH3-TPD 譜圖

表2 催化劑的Py-FT-IR 結果
2.2 FCC 柴油的加氫改質反應性能
2.2.1 3 種催化劑的加氫改質反應性能
在催化劑制備方法和金屬Pt 負載量相同、且3種催化劑的孔道結構相似的情況下,分子篩載體的酸性特征將決定預精制FCC 柴油加氫改質反應性能。在固定床連續加氫反應裝置上,詳細考察了SRO-1、SRO-2 和SRO-3 催化劑的酸性對加氫預精制FCC 柴油中芳烴飽和與選擇性開環反應性能的影響。表3 是在反應溫度300℃、壓力6.0MPa、液時空速4.0h-1和H2/oil 體積比750∶1 的條件下SRO-1、SRO-2 和SRO-3 催化劑上預精制FCC 柴油加氫改質反應性能數據。由表3 可知,與預精制FCC柴油族組成相比,SRO-1 催化劑上加氫改質反應得到的柴油餾分中芳烴質量分數顯著降至4.2%,脫芳率達到了92.0%,其中芳烴飽和率為71.7%,芳烴開環率為20.3%,十六烷值提高了10.5 個單位;SRO-2 催化劑上加氫改質反應得到的柴油餾分中芳烴質量分數也降至4.2%,脫芳率達到了92.0%,芳烴飽和率為76.5%,芳烴開環率為15.5%,十六烷值提高了15.1 個單位;SRO-3 催化劑上加氫改質反應得到的柴油餾分中芳烴質量分數降至28.0%,脫芳率達到了46.8%,其中芳烴飽和率為40.7%,芳烴開環率為6.1%,十六烷值提高了8.2 個單位。

表3 不同催化劑的加氫改質反應性能
上述數據表明,SRO-1 催化劑和SRO-2 催化劑在脫芳率相同情況下,前者芳烴開環率高了4.8 個百分點,而十六烷值增幅卻小了4.6 個單位。這是因為對于酸催化反應來說,酸量越大,酸強度越高,反應活性越高,過度裂化性能越強[15]。與SRO-2催化劑相比,SRO-1 催化劑的酸強度與酸量較高,在進行開環反應的同時,SRO-1 催化劑會帶來更多的裂化反應,生成更多的小分子烴類[16-17],相應就會降低產品的十六烷值(如n-C12的十六烷值為87,n-C15的十六烷值為96,n-C16的十六烷值為100,正十二烷基苯的十六烷值為68,正十四烷基苯的十六烷值為72,對于直鏈烷烴,分子中每相差1 個碳原子,十六烷值相差3~4 個單位;對于烷基苯,其烷基側鏈每相差1 個碳原子,十六烷值相差約2 個單位[5])。SRO-3 催化劑的脫芳率較SRO-2 催化劑減少了45.2 個百分點,且芳烴飽和反應占主導,十六烷值增幅較小,這是由于SRO-3 催化劑的酸強度與酸量均低于SRO-2 催化劑,酸量越小,酸強度越低,反應活性越低,芳烴選擇性開環反應性能越差,主要發生芳烴飽和反應,相應十六烷值的增加主要由生成的環烷烴貢獻。
圖4 是柴油餾分的模擬蒸餾曲線。由圖4 可知,與預精制FCC 柴油的餾程比較,SRO-1 催化劑上加氫改質反應得到的液體產品的餾程前移約13 個百分點,SRO-2 催化劑上液體產品的餾程前移約8 個百分點,SRO-3 催化劑上液體產品的餾程前移約3個百分點。上述數據顯示,SRO-1、SRO-2 和SRO-3催化劑上產物中的輕組分依次減少。這說明催化劑的酸性越強,其裂化反應性能越強,從而導致產物中輕組分越多。因SRO-1 催化劑的酸性最強,故其產物中輕組分含量最多;SRO-3 催化劑的酸性最弱,其產物中輕組分含量最少。此結果也進一步證明在芳烴選擇性開環大幅度提高柴油十六烷值過程中, 高活性和高選擇性的加氫改質催化劑的酸量和酸強度要適中,酸性太強易發生過度裂化反應,酸性太弱反應活性差,強酸和弱酸均對十六烷值提高幅度有限。

圖4 柴油餾分的模擬蒸餾曲線
2.2.2 使用SRO-2 催化劑時反應條件對芳烴含量和十六烷值的影響
反應條件對劣質柴油加氫改質反應非常重要,采用具有較好加氫反應活性和芳烴選擇性開環反應性能的SRO-2 催化劑,以預精制FCC 柴油為原料,進一步考察了不同反應條件對脫除芳烴和提高十六烷值的影響。圖5 是SRO-2 催化劑在不同反應溫度和不同反應壓力條件下,加氫脫芳和提高十六烷值的反應性能曲線。由圖5 可知,SRO-2 催化劑在低溫下(240℃)就有較好的加氫脫芳能力,隨著反應溫度不斷升高,其脫芳性能逐漸增強,十六烷值不斷提高,當反應溫度達到300℃時,產物中芳烴質量分數已低于10%,十六烷值大于55,達到了國V柴油質量要求,這證明貴金屬催化劑在較低反應溫度下就有較高的加氫飽和性能,隨著反應溫度的升高,其裂化性能逐漸加強,環烷烴選擇性開環裂化為鏈烷烴的概率增加,從而大幅度提高了柴油餾分的十六烷值。低壓不利于芳烴的加氫反應,隨著反應壓力的不斷提高,SRO-2 催化劑的加氫脫芳能力逐漸增強,柴油十六烷值不斷提高,證明提高反應壓力有利于芳烴飽和反應和選擇性開環反應。在反應溫度 300℃時,反應壓力從 4.0MPa 提高到8.0MPa,柴油產品的芳烴含量和十六烷值變化很小,其柴油產品質量均能達到國V 標準,因此綜合考慮裝置投資、操作成本、產品質量以及與加氫預精制段操作條件的匹配,加氫改質段操作壓力適宜在6.0~8.0MPa。
柴油加氫改質主要涉及3 個化學反應:芳烴飽和反應、芳烴選擇性開環反應、裂化反應。液時空速會影響預精制FCC 柴油在加氫改質催化劑上的停留時間,進而會影響上述3 個反應。十六烷值的大小主要是由芳烴選擇性開環反應決定,圖6 是SRO-2 催化劑在不同空速條件下加氫脫芳和提高十六烷值的反應性能曲線。由圖6 可知,隨著反應空速的增大,SRO-2 催化劑脫芳能力逐漸下降,十六烷值增幅不斷降低,這是由于液時空速增大,預精制FCC 柴油在SRO-2 催化劑上的停留時間縮短,芳烴飽和與開環裂化能力減弱,故產品中芳烴含量高,十六烷值低。隨著反應空速的降低,芳烴飽和反應增加,然后進一步發生選擇性開環反應,同時伴隨著裂化反應的增加,生成低十六烷值的小分子烴相應增加,從而影響了十六烷值的提高(圖1),所以芳烴含量變化較大,十六烷值變化不明顯。大空速條件下,若要使加氫改質柴油滿足國V 標準,必須適當提高反應溫度,以增加芳烴飽和反應和開環裂化反應,以此降低產品中芳烴含量,提高十六烷值。如果反應溫度過高,裂化反應加劇,產品中輕組分含量增加,目標產品收率就會降低。因此,綜合考慮柴油產品收率和質量,加氫改質反應的空速適宜為2.0~4.0h-1。

圖5 反應溫度和壓力對芳烴含量和十六烷值的影響

圖6 液時空速對芳烴含量和十六烷值的影響
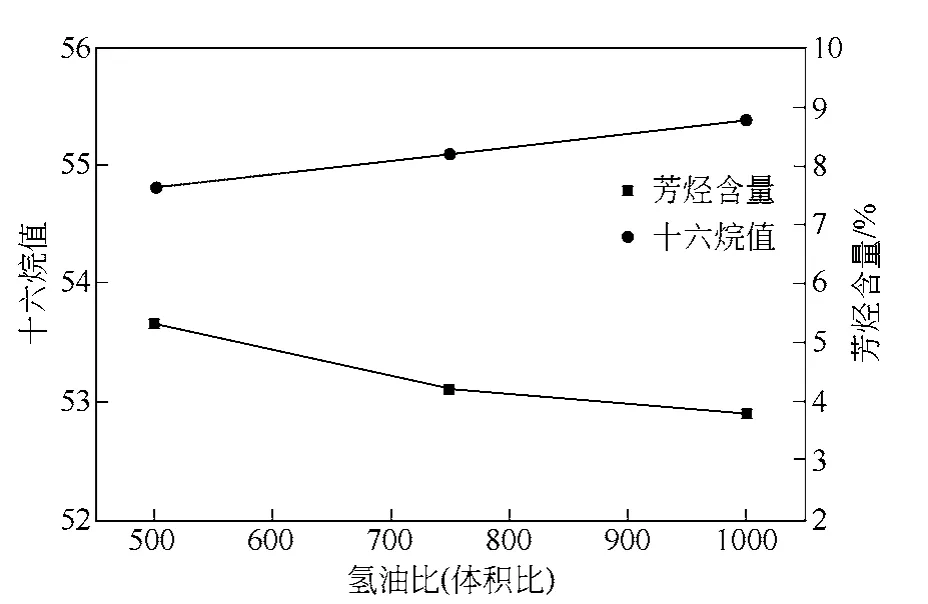
圖7 H2/oil 體積比對芳烴含量和十六烷值的影響
圖7 是SRO-2 催化劑在不同氫油體積比條件下 加氫脫芳和提高十六烷值的反應性能曲線。由圖7可知,在H2/oil 體積比為500~1000 時,SRO-2 催化劑的脫芳能力和加氫改質柴油的十六烷值增幅變化不大。
綜上可知,以預精制FCC 柴油為原料生產滿足國Ⅴ標準的柴油時,為了提高柴油加氫改質反應單元的原料加工量和降低操作成本,采用SRO-2 催化劑進行加氫改質反應的較佳工藝條件為:反應溫度300℃,反應壓力6.0MPa,液時空速4.0h-1,H2/oil體積比750。
3 結 論
在0.5%Pt/USY、0.5%Pt/1.0K-USY 和0.5%Pt/ 2.0K-USY 催化劑的金屬Pt 負載量相同以及孔道結構相似前提下,以預精制FCC 柴油為原料,經加氫改質反應生產滿足國V 質量標準的清潔柴油時,催化劑的酸性起了非常重要的作用,酸密度和酸強度分布決定了催化劑的活性和選擇性。具有較高的加氫活性和適宜酸性的催化劑更有利于加氫改質反應,避免過多裂化反應的發生,在改善柴油產品質量的同時提高了柴油產品的收率。通過對預精制FCC 柴油進行加氫改質反應性能研究,得知0.5%Pt/1.0K-USY 催化劑具有較好的加氫活性以及較高的加氫脫芳和提高十六烷值的能力,在反應溫度300℃、反應壓力6.0MPa、液時空速4.0h-1、H2/oil體積比750 條件下,預精制FCC 柴油經加氫改質反應,柴油收率為95.1%(質量分數),柴油餾分中芳烴質量分數為4.2%,十六烷值為55.1,達到了國V柴油質量標準。 0.5%Pt/USY 催化劑與0.5%Pt/1.0K-USY 催化劑相比,在脫芳率相同和芳烴開環率高了4.8 個百分點的情況下,十六烷值增幅卻少了4.6 個單位,這是0.5%Pt/USY 催化劑較強的酸性使得芳烴在開環反應的同時發生了較多的裂化副反應,生成了更多的小分子烴類,從而降低了柴油的收率和十六烷值;0.5%Pt/2.0K-USY 催化劑比0.5%Pt/1.0K-USY 催化劑的脫芳率減少了45.2 個百分點,這是0.5%Pt/2.0K-USY 催化劑的較弱酸性使得加氫反應活性降低,芳烴選擇性開環反應性能變差,十六烷值的增加主要由芳烴飽和生成的環烷烴貢獻。
[1] Gutiérrez A,Arandes J M,Casta?o P,et al. Enhancement of aromatic hydro-upgrading on a Pt catalyst by promotion with Pd andshape-selective supports[J]. Fuel Processing Technology,2012,101:64-72.
[2] 王甫村,朱金玲,田然,等. 用于FCC 柴油加氫改質的選擇性開環催化劑研究進展[J]. 化工進展,2011,30(9):1951- 1955.
[3] Heckel T,Thakkar V,Behraz E,et al. NPRA Annual Meeting. San Francisco,1998[C]// National Petrochemical & Refiners Association,Washington,1998.
[4] Eng O T,Kennedy J E. NPRA Annual Meeting. San Antonio[C]// National Petrochemical & Refiners Association,Washington,2000.
[5] Santana R C,Do P T,Santikunaporn M,et al. Evaluation of different reaction strategies for the improvement of cetane number in diesel fuels[J]. Fuel,2006,85(5-6):643-656.
[6] Cooper B H,Donnis B B L. Aromatic saturation of distillates:An overview[J]. Applied Catalysis A:General,1996,137(2):203-223.
[7] Mcvicker G B,Daage M,Touvelle M S,et al. Selective ring opening of naphthenic molecules[J]. Journal of Catalysis,2002,210(1):137-148.
[8] Knottenbelt C. Mossgas “gas-to-liquid” diesel fuels-an environmentally friendly option[J]. Catalysis Today,2002,71(3-4),437-445.
[9] Song C,Ma X. New design approaches to ultra-clean diesel fuels by deep desulfurization and deep dearomatization[J]. Applied Catalysis B:Environmental,2003,41(1-2):207-238.
[10] Gutiérrez A,Arandes J M,Casta?o P,et al. Effect of space velocity on the hydrocracking of light cycle oil over a Pt-Pd/HY zeolite catalyst[J]. Fuel Processing Technology,2012,95:8-15.
[11] Calemma V,Ferrari M,Rabl S,et al. Selective ring opening of naphthenes:From mechanistic studies with a model feed to the upgrading of a hydrotreated light cycle oil[J]. Fuel,2013,111:763-770.
[12] D’Ippolito S A,Gutierrez L B,Vera C R,et al. Pt-Mg-Ir/Al2O3and Pt-Ir/HY zeolite catalysts for SRO of decalin. Influence of Ir content and support acidity[J]. Applied Catalysis A:General,2013,452:48-56.
[13] Gutiérrez A,Arandes J M,Casta?o P,et al. Preliminary studies on fuel production through LCO hydrocracking on noble-metal supported catalysts[J]. Fuel,2012,94:504-515.
[14] D’Ippolito S A,Especel C,Vivier L,et al. Influence of the Br?nsted acidity,SiO2/Al2O3ratio and Rh-Pd content on the ring opening:Part I. Selective ring opening of decalin[J]. Applied Catalysis A:General,2014,469:532-540.
[15] H?chtl M,Jentys A,Vinek H. Acidity of SAPO and CoAPO molecular sieves and their activity in the hydroisomerization of n-heptane[J]. Microporous and Mesoporous Materials,1999,31(3):271-285.
[16] Arve K,M?ki-Arvela P,Er?nen K,et al. Utilisation of a multitubular reactor system for parallel screening of catalysts for ring opening of decalin in continuous mode[J]. Chemical Engineering Journal,2014,238:3-8.
[17] Tailleur R G. Deactivation of WNiPd/TiO2Al2O3catalyst during the upgrading of LCO[J]. Fuel,2008,87(12):2551-2562.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
