不同晶相結構二氧化錳催化完全氧化乙醇
2015-08-15 08:33:52張江浩張長斌中國科學院生態環境研究中心大氣污染控制中心環境模擬與污染控制國家聯合重點實驗室北京100085
物理化學學報 2015年2期
關鍵詞:催化劑
張 潔 張江浩 張長斌 賀 泓(中國科學院生態環境研究中心大氣污染控制中心,環境模擬與污染控制國家聯合重點實驗室,北京100085)
不同晶相結構二氧化錳催化完全氧化乙醇
張潔張江浩張長斌賀泓*
(中國科學院生態環境研究中心大氣污染控制中心,環境模擬與污染控制國家聯合重點實驗室,北京100085)
采用水熱合成法制備了α-MnO2、β-MnO2、γ-MnO2和δ-MnO2催化劑,運用N2吸脫附實驗、X射線衍射(XRD)、X射線光電子能譜(XPS)和H2程序升溫還原(H2-TPR)等方法對催化劑進行了表征,并將催化劑用于催化完全氧化乙醇反應中,考察了不同晶型MnO2催化劑催化氧化乙醇活性的差異,探討了催化劑晶型結構與催化氧化活性的關聯.結果表明,不同晶型的MnO2催化劑催化氧化乙醇活性差異顯著,活性順序為α-MnO2>δ-MnO2>γ-MnO2>β-MnO2.系列表征結果顯示,晶體結晶度和比表面積不是影響不同晶型MnO2催化劑活性的主要原因,α-MnO2催化劑具有的較高晶格氧濃度和較高的可還原性是其具有良好催化氧化乙醇活性的關鍵因素.
MnO2催化劑;低溫催化氧化;乙醇完全氧化;揮發性有機物;晶格氧
www.whxb.pku.edu.cn
1 引言
揮發性有機物(VOCs)是大氣污染的主要來源之一,是大氣細粒子(PM2.5)的主要前驅物之一,1降低VOCs的排放對保護人類健康和生態環境具有重要意義.乙醇作為一種常見的揮發性有機物,存在于以乙醇為混合燃料的機動車尾氣中,乙醇混合汽油為動力的機動車尾氣中釋放的乙醇氣體及其部分氧化產物乙醛,對人體健康具有直接危害.2,3隨著乙醇添加燃料在我國的推廣普及,針對機動車尾氣中乙醇污染物高效去除手段的研究具有重要的環境意義和社會價值.
催化氧化法因具有轉化率高,起燃溫度低,工藝簡單,節約能源和無二次污染等優點,已成為廣泛應用的醇類污染物去除手段.現有催化氧化方法中使用的催化劑主要分為兩大類:貴金屬催化劑和金屬氧化物催化劑.在貴金屬催化劑中,Pt、Pd、Au、Ru已被研究作為氧化乙醇的催化劑;4-6金屬氧化物催化劑中,Cu、Cr、V、Co、Ce、Mn氧化物是常用的乙醇等有機污染物氧化催化劑.7-13目前,某些金屬氧化物催化劑已經可以達到和貴金屬催化劑相近的低溫催化氧化活性,Wang和Li10報道了以Mn-SO4為前驅體制備的OMS-2型錳氧化物催化劑催化乙醇完全氧化為CO2的溫度僅為160°C,活性優于貴金屬催化劑.金屬氧化物催化劑使用成本低廉并具有很好的抵抗反應中有毒物質的能力.基于以上優點,關于金屬氧化物催化劑用于催化完全氧化乙醇的研究具有很高的現實意義.
在眾多氧化物催化劑中,錳氧化物具有優異的催化氧化活性,越來越多的學者將研究重點轉向此處,以期研制出更為高效實用的催化劑.14-17其中MnO2催化劑是近期的研究熱點,它由[MnO6]八面體基元通過不同排列方式形成不同的晶型結構,如α-、β-、γ-、δ-MnO2等,18-21晶格結構和晶胞參數不同,催化性能也存在很大差別.22-25Liang等22研究了α-、β-、γ-、δ-MnO2催化劑CO氧化活性的差異,發現催化劑對CO的吸附能力、Mn―O鍵強度和Mn物種的氧化還原過程是影響催化劑活性的主要原因.Xu等23制備了α-、β-MnO2為載體的Ag-MnO2催化劑,通過對比催化劑CO氧化性能,發現晶體結構和表面結構是影響催化劑活性的主要因素.戴韻等24制備了α-、δ-MnO2,并考察了其低溫NH3選擇性催化還原(NH3-SCR)NOx的性能,研究發現比表面積不是影響兩種MnO2催化劑活性的主要因素,影響活性的主要因素是催化劑的晶相結構和表面性質.李經緯等25采用水熱法分別合成了α-、β-、γ-MnO2納米棒,考察了其催化氧化氯苯的能力差異,發現三種晶型MnO2納米棒中氧物種的可還原能力與晶格氧的相對含量是影響其催化氧化活性的主要因素.上述研究都發現了晶型的差異會導致MnO2催化劑活性的差別,因此進行不同晶型MnO2催化乙醇性能的研究對尋找和制備具有更高催化性能的錳氧化物催化劑具有重要意義.
本文通過水熱合成法制備了α-、β-、γ-、δ-MnO2催化劑,比較了它們催化乙醇氣體完全氧化的活性,探索了晶體結構對催化劑乙醇完全氧化活性的影響.
2 實驗部分
2.1催化劑的制備
利用水熱合成法制備不同晶型的MnO2催化劑.10,22α-MnO2,0.525 g MnSO4·H2O(國藥集團化學試劑有限公司,分析純)和1.25 g KMnO4(國藥集團化學試劑有限公司,分析純)混合于80 mL去離子水中,添加2 mL濃HNO3(國藥集團化學試劑有限公司,分析純),磁力攪拌1 h,將混合溶液移至100 mL的特氟龍反應釜中,100°C水熱反應24 h.β-MnO2,1.69 g MnSO4·H2O(國藥集團化學試劑有限公司,分析純)和2.28 g(NH4)2S2O8(天津市福晨化學試劑廠,分析純)混合于80 mL去離子水中,攪拌1 h,將混合溶液移至100 mL的特氟龍反應釜中,140°C水熱反應12 h.γ-MnO2,3.375 g MnSO4·H2O(國藥集團化學試劑有限公司,分析純)和4.575 g (NH4)2S2O8(天津市福晨化學試劑廠,分析純)混合于80 mL去離子水中,攪拌1 h,將混合溶液移至100 mL的特氟龍反應釜中,90°C水熱反應24 h.δ-MnO2,0.275 g MnSO4·H2O(國藥集團化學試劑有限公司,分析純)和1.5 g KMnO4(國藥集團化學試劑有限公司,分析純)混合于80 mL去離子水中,攪拌1 h,將混合溶液移至100 mL的特氟龍反應釜中,240°C水熱反應24 h.19將以上反應得到的固體氧化物多次洗滌至中性,置于100°C干燥過夜,得到固體樣品經400°C焙燒2 h后研磨至40-60目備用.
2.2催化劑的表征
N2吸附-脫附實驗使用美國Quantachrome公司的Autosorb-iQ-1MP自動吸附裝置,在-196°C(77 K)溫度下,用液氮吸附法測定.測量前,將樣品在氮氣流中100°C持續加熱1 h以除去樣品表面吸附水,然后在300°C持續4 h真空脫氣.測定了催化劑的比表面積,孔容和孔徑.粉末X射線衍射(XRD)分析使用瑞士Bruker公司D8-ADVANCE射線衍射儀,入射光源為Cu Kα輻射,管電壓40 kV,管電流40 mA,掃描范圍10°-90°,掃描速率4(°)·min-1.實驗采用日本ULVAC-PHI公司的PHI Quantera型掃描X射線光電子能譜儀對樣品進行分析.采用Al Kα(1486.7 eV)作為X射線靶,所有元素的結合能用C 1s(284.8 eV)校正電荷位移,掃描步長為0.1 eV.程序升溫還原(H2-TPR)測試采用美國Micro公司Auto Chem II 2920化學吸附儀,以10%(體積分數,φ)H2/ Ar混合氣為還原氣,樣品用量為30 mg,以10°C· min-1的速率升溫至900°C.
2.3催化劑的評價
在自制的石英管微型固定床反應器中評價了催化劑的乙醇氧化反應性能.反應過程中采用的乙醇濃度為800×10-6(φ),O2濃度為20%(φ),N2為平衡氣,反應氣總流量為100 mL·min-1.催化劑用量為50 mg,為消除內外擴散影響,采用40-60目的催化劑進行評價,空速(GHSV)約為36000 h-1.40-220° C,每隔10或20°C,采集2-3個數據點,平衡時間為1.5 h.采用氣相色譜Agilent 7890(美國安捷倫公司)對產物進行檢測,氣相色譜配有Porapak-Q色譜柱,采用熱導檢測器(TCD)檢測乙醇、乙醛和CO2等產物.碳平衡達到100%±5%.
乙醇轉化率(X),乙醛和二氧化碳(CO2)生成率(Y)分別由以下公式計算而得:

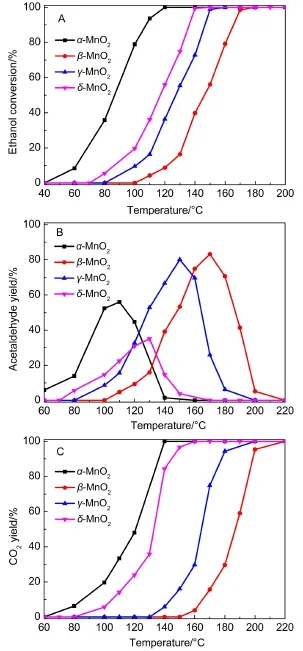
圖1 不同晶型的MnO2催化劑氧化乙醇反應的乙醇轉化率(A),乙醛生成率(B)和CO2生成率(C)Fig.1 Ethanol conversion(A),acetaldehyde yield(B),and CO2yield(C)of MnO2catalysts with different phase structures during ethanol oxidation reactionreaction conditions:800×10-6(volume fraction,φ)ethanol,20%(φ)O2,N2as balance gas;gaseous hourly space velocity(GHSV):36000 h-1
3 結果與討論
3.1不同晶型MnO2氧化乙醇活性
不同晶型的MnO2氧化物催化氧化乙醇的活性如圖1所示.四種晶型MnO2催化劑對乙醇均有較高的催化氧化活性,特別是低溫活性遠高于相關文獻報道的,包括貴金屬負載型催化劑、4-6錳氧化物、26Cu-Mn27和Mn-Ce28催化劑.從圖1A中可以看出,不同晶型的MnO2氧化物催化劑對乙醇氧化反應活性具有很大差異,乙醇氧化活性排序為α-MnO2>δ-MnO2>γ-MnO2>β-MnO2.α-MnO2催化劑活性最優,在120°C即可將反應氣中的乙醇氣體完全轉化;β-MnO2催化劑活性最差,其完全轉化乙醇的溫度為180°C.如圖1B所示,δ-MnO2催化劑氧化乙醇氣體反應中,有害副產物乙醛的生成率最低,最高產率小于30%;γ-、β-MnO2乙醇氧化反應過程中副產物乙醛的生成率較高,最高達80%,對于多步氧化反應而言,反應物或中間產物在催化劑表面的強吸附可能會占據催化劑的活性位,從而降低反應活性,因此較高的乙醛生成率導致了其乙醇完全氧化活性大大降低.如圖1C所示,α-、β-、γ-、δ-MnO2催化劑將乙醇完全氧化為CO2的溫度分別為140,220,200,160°C.可以看出γ-和δ-MnO2兩種催化劑二氧化碳生成率的差別與兩者乙醇轉化率的差別相比更大,說明雖然乙醇的轉化率在γ-和δ-MnO2上幾乎一樣,但是在γ-MnO2催化劑上,更多的乙醇被部分氧化生成乙醛,從而導致兩者完全氧化乙醇為CO2的活性存在較大差異.β-MnO2同樣有大量乙醇被部分氧化生成乙醛,導致在該催化劑上乙醇完全氧化為CO2的溫度遠遠高于其它三種晶型的MnO2催化劑.
3.2物理性質與晶體結構
不同晶相MnO2催化劑的XRD圖如圖2所示. 圖2a在2θ為12.7°、18.0°、25.5°、37.4°、41.8°、52.0°、55.3°和60.0°處分別出現對應α-MnO2四方晶相(110)、(200)、(310)、(211)、(301)、(411)、(600)和(521)晶面的衍射峰,氧化物的晶型結構完全符合α-MnO2結構(JCPDS 44-0141).圖2b在2θ為28.6°、37.3°、41.0°、42.8°、56.7°和59.4°處分別出現對應β-MnO2的(110)、(101)、(200)、(111)、(211)和(220)晶面的衍射峰,衍射峰歸屬于β-MnO2的四方晶系(JCPDS 24-0735).圖2c在2θ為22.3°、37.1°、42.3°和56.9°處分別出現對應γ-MnO2的(120)、(131)、(300)和(160)晶面的衍射峰,歸屬為γ-MnO2(JCPDS 14-0644).圖2d在 2θ為12.3°、25.1°和37.2°處分別出現對應δ-MnO2的(001)、(002),和(111)晶面衍射峰,歸屬為δ-MnO2(JCPDS 80-1098)結構.與α-、β-MnO2相比,γ-、δ-MnO2的XRD的衍射峰強度低,峰形展寬大,表明γ-、δ-MnO2這兩種氧化物的結晶度較低.對比這四種晶型MnO2催化劑催化氧化乙醇性能的結果,發現結晶度對催化劑活性的影響效果不顯著,李經緯等25的研究中也發現,MnO2催化劑的結晶程度不是影響其活性的主要原因.

圖2 不同晶相MnO2催化劑的XRD譜圖Fig.2 XRD patterns of MnO2catalysts with different phase structures(a)α-MnO2,(b)β-MnO2,(c)γ-MnO2,(d)δ-MnO2
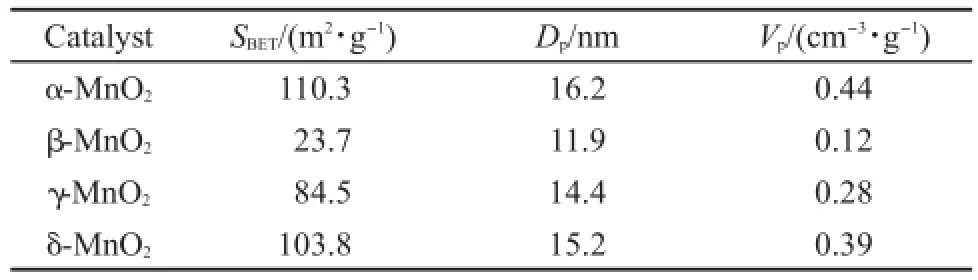
表1 不同晶相MnO2催化劑的物理性質Table 1 Physical properties of MnO2catalysts with different phase structures
表1是四種催化劑樣品通過N2-吸附-脫附實驗得到的催化劑樣品比表面積(SBET),平均孔徑(DP)和總孔容(VP)等孔結構參數.結果表明,四種不同晶型MnO2催化劑的比表面積差異顯著,其比表面積變化規律為α-MnO2>δ-MnO2>γ-MnO2>β-MnO2.
由于不同晶型的MnO2比表面積差異顯著,為了扣除比表面積對催化劑活性的影響,在110°C條件下,不同MnO2催化劑的比活性進行了單位面積歸一化換算研究,結果列于表2中.其中,計算所用的比表面積為各個MnO2催化劑根據BET方法計算所得的表面積.可以看出歸一化之后,催化劑在110°C時的催化活性順序為α-MnO2>δ-MnO2>γ-MnO2>β-MnO2,說明催化劑的比表面積不是影響MnO2催化劑活性的主要因素.
3.3不同晶型MnO2催化劑的H2-TPR分析
采用H2-TPR方法對不同催化劑可還原性進行了表征,結果如圖3所示.α-、β-、γ-、δ-MnO2催化劑H2氣還原峰峰型具有很大差異.α-MnO2氧化物在280和310°C分別出現兩個H2消耗峰,經過分峰處理得到此兩處的氫氣消耗量分別為6.87和3.79 mmol·g-1,與理論上還原過程中H2消耗量比例(2:1)相近,表明這兩個還原過程分別對應了MnO2→Mn3O4和Mn3O4→MnO.24,25β-MnO2氧化物的第一個主要還原峰出現在290°C,該還原過程由兩部分疊加而成,通過分峰計算,其第一峰氫氣總消耗量為7.41 mmol·g-1,第二個還原峰出現在400°C,氫氣消耗量為3.71 mmol·g-1,同樣對應于MnO2→Mn3O4→MnO的還原過程,其高溫還原峰出現溫度遠大于α-MnO2.γ-MnO2催化劑的還原峰與β-MnO2相似,但向低溫方向偏移,分別位于280和380°C處.δ-MnO2氧化物具有總包H2消耗峰,根據峰型可分成3個疊加峰,分別出現在280、293和320°C,經過分峰處理得到此三處的氫氣消耗量分別為5.4、1.9和4.2 mmol·g-1,分別對應了MnO2→Mn2O3,Mn2O3→Mn3O4和Mn3O4→MnO,這與葉青等31的研究結果相似.四種晶型催化劑的H2-TPR實驗結束后樣品均變為綠色,總氫氣消耗量在10.66-11.48 mmol·g-1之間,H/Mn摩爾比均約等于2,這對應了MnO2還原至MnO的消耗量,說明四種晶型MnO2催化劑氫氣還原的最終產物均MnO,也與樣品最終形態匹配.
催化劑還原峰出現的溫度范圍越低,其可被還原的能力就越強,催化劑氧物種的流動性越好.因此,β-MnO2中氧物種的流動性要差于其他三種MnO2催化劑.值得注意的是,與δ-MnO2相比,α-MnO2的低溫峰與δ-MnO2相近,但高溫峰遠低于δ-MnO2.α-MnO2的還原過程高度集中于250-320°C這個溫度范圍內,在320°C左右即完全還原為MnO,無論是還原溫度區間還是還原終止溫度均低于其它晶型的MnO2,因此α-MnO2催化劑中氧物種的流動性最優.綜上所述,不同晶型MnO2催化劑氧物種流動性從優到劣的次序為α-MnO2>δ-MnO2>γ-MnO2>β-MnO2.
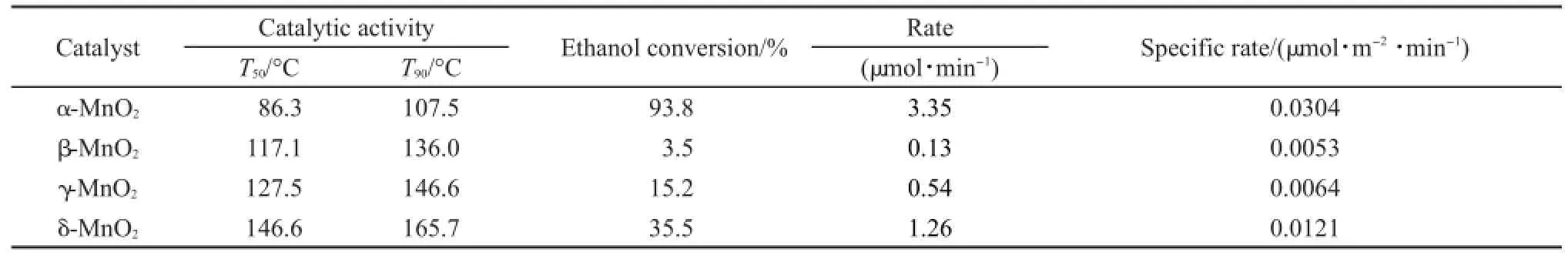
表2 MnO2催化劑上乙醇氧化T50,T90和單位表面積轉化率Table 2 T50,T90,and specific rate of ethanol oxidation over the MnO2catalysts
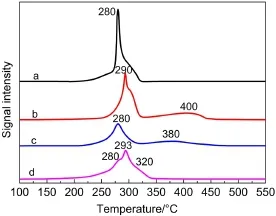
圖3 MnO2催化劑的H2-TPR圖Fig.3 H2-TPR profiles of MnO2catalysts(a)α-MnO2,(b)β-MnO2,(c)γ-MnO2,(d)δ-MnO2
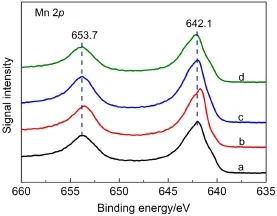
圖4 MnO2催化劑中Mn 2p的XPS圖Fig.4 Mn 2p XPS spectra of MnO2catalysts(a)α-MnO2,(b)β-MnO2,(c)γ-MnO2,(d)δ-MnO2
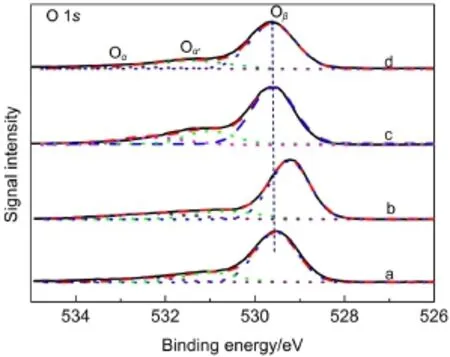
圖5 MnO2催化劑中O 1s的XPS圖Fig.5 O 1s XPS spectra of MnO2catalysts(a)α-MnO2,(b)β-MnO2,(c)γ-MnO2,(d)δ-MnO2
3.4不同晶型MnO2催化劑的XPS分析
圖4和圖5為不同晶型MnO2催化劑Mn 2p和O 1s的XPS圖譜,用Peakfit軟件對O 1s譜圖進行了擬合,并計算了氧物種Oα、Oα?和Oβ的摩爾比例,結果匯總于表3.從圖4中可以看出,α-、β-、γ-、δ-MnO2催化劑中Mn 2p2/3特征峰的結合能都位于642.1 eV左右,表明四種晶型MnO2中錳元素主要為Mn4+,25不同晶型MnO2催化劑表面Mn的價態相同.
由圖5知,經過對O 1s分峰擬合,可將氧物種分為三種類型:Oβ、Oα和Oα'.結合能位于529.3-529.7 eV的氧物種Oβ歸屬于MnO2的晶格氧(O2-)31;結合能位于531.0-531.4 eV的氧物種Oα?歸屬于MnO2的表面氧(如O-、OH-、CO23-等);結合能位于532.4-532.8 eV左右的氧物種Oα歸屬于MnO2吸附的水分子.32從表3可知,α-、β-、γ-、δ-MnO2催化劑中晶格氧含量Oβ/(Oα+Oα?+Oβ)的摩爾比值依次為78.7%、69.5%、70.5%、75.5%,在這四種MnO2催化劑中,β-MnO2的晶格氧占總的氧物種的比例最低,α-MnO2的晶格氧含量最高,晶格氧相對含量的順序為α-MnO2>δ-MnO2>γ-MnO2>β-MnO2.α-MnO2的晶格氧濃度高于其它三種催化劑,這與乙醇催化氧化反應中得到的活性數據相吻合.表面積歸一化的活性結果中,γ-、β-MnO2催化劑單位比表面上的轉化率相近,與兩個晶格氧含量相近的結果吻合.在錳氧化物催化劑催化氧化醇類的反應中,催化劑的晶格氧是影響催化劑的重要因素,晶格氧含量高的催化劑其活性更好.17,33因此,在系列不同晶型催化劑中,α-MnO2催化劑具有最高的晶格氧含量是其具有最高反應活性的重要因素之一.
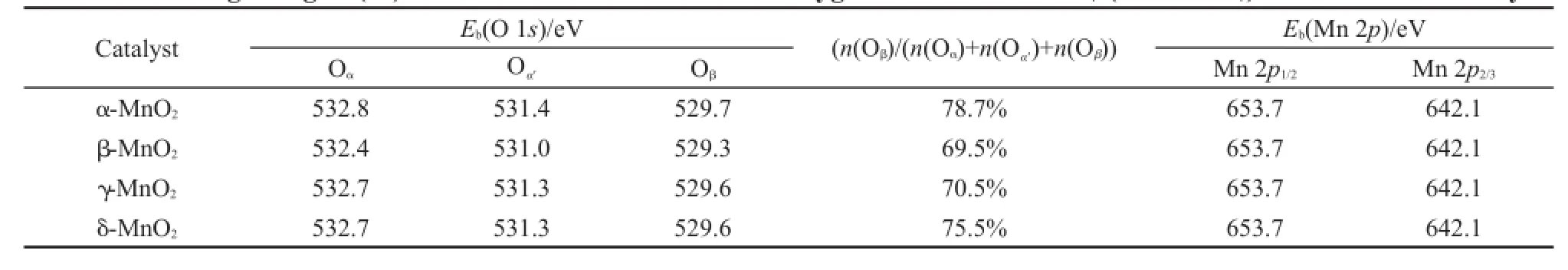
表3 MnO2催化劑的表面原子結合能(Eb)及晶格氧Oβ/(Oα+Oα'+Oβ)的摩爾比Table 3 Binding energies(Eb)of surface elements and lattice oxygen molar ratios of Oβ/(Oα+Oα'+Oβ)over the MnO2catalysts
4 結論
本文用水熱合成法制備了具有不同晶型結構的α-MnO2、β-MnO2、γ-MnO2、δ-MnO2催化劑,考察了此類MnO2催化劑催化完全氧化乙醇的性能,并對催化劑進行了系列表征.結果表明,MnO2催化劑的晶體結構對其乙醇氧化活性具有重要影響;α-MnO2催化劑具有最優的低溫乙醇氧化性能,乙醇完全轉化的溫度為120°C,并在140°C將乙醇完全氧化生成二氧化碳,β-MnO2催化劑的活性最差,乙醇完全氧化的溫度高達220°C;通過比較催化劑的比表面積反應速度,可以看出不同晶型的MnO2催化劑的比表面積對反應活性影響不大.催化劑的活性主要受晶格氧含量和其流動性的影響.
References
(2)Grosjean,D.;Grosjean,E.;Gertler,A.W.Environ.Sci.Technol. 2001,35,45.doi:10.1021/es001326a
(3)Jacobson,M.Z.Environ.Sci.Technol.2007,41,4150.doi:10.1021/es062085v
(4)Avgouropoulos,G.;Oikonomopoulos,E.;Kanistras,D.;Ioannides,T.Appl.Catal.B:Environ.2006,65,62.doi:10.1016/j.apcatb.2005.12.016
(5)Cordi,E.M.;Falconer,J.L.J.Catal.1996,162,104.doi:10.1006/jcat.1996.0264
(6)Mitsui,T.;Tsutsui,K.;Matsui,T.;Kikuchi,R.;Eguchi,K.Appl. Catal.B:Environ.2008,81,56.doi:10.1016/j. apcatb.2007.12.006
(7)Idriss,H.;Seebauer,E.G.J.Mol.Catal.A 2000,152,201.doi:10.1016/S1381-1169(99)00297-6
(8)Li,H.J.;Tana;Zhang,X.J.;Huang,X.M.;Shen W.J.Catal. Commun.2011,12,1361.doi:10.1016/j.catcom.2011.05.016
(9)Ye,Q.;Gao,Q.;Zhang,X.R.;Xu,B.Q.Catal.Commun.2006,7,589.doi:10.1016/j.catcom.2006.01.023
(10)Wang,R.H.;Li,J.H.Environ.Sci.Technol.2010,44,4282. doi:10.1021/es100253c
(11)Zhao,P.;Wang,C.N.;He,F.;Liu,S.T.RSC Adv.2014,4,45665.doi:10.1039/C4RA07843H
(12)Liang,S.H.;Teng,F.;Yao,W.Q.;Zhu,Y.F.Acta Phys.-Chim. Sin.2008,24,205.[梁淑惠,滕飛,姚文清,朱永法.物理化學學報,2008,24,205.]doi:10.3866/PKU.WHXB20080205
(13)Li,J.W.;Zhao,P.;Liu,S.T.Appl.Catal.A:Gen.2014,482,363.doi:10.1016/j.apcata.2014.06.013
(14)Tang,X.F.;Li,Y.G.;Huang,X.M.;Xu,Y.D.;Zhu,H.Q.;Wang,J.G.;Shen,W.J.Appl.Catal.B:Environ.2006,62,265.doi:10.1016/j.apcatb.2005.08.004
(15)Morales,M.R.;Barbero,B.P.;Cadús,L.E.Appl.Catal.B:Environ.2007,74,1.doi:10.1016/j.apcatb.2007.01.008
(16)Njagi,E.C.;Chen,C.H.;Genuino,H.;Galindo,H.;Huang,H.;Suib,S.L.Appl Catal.B:Environ,2010,99,103.doi:10.1016/j. apcatb.2010.06.006
(17)Luo,J.;Zhang,Q.H.;Garcia-Martin z,J.;Suib,S.L.J.Am. Chem.Soc.2008,130,3198.doi:10.1021/ja077706e
(18)Ye,Q.;Zhao,J.S.;Huo,F.F.;Wang,D.;Cheng,S.Y.;Kang,T. F.;Dai,H.X.Microporous Mesoporous Mat.2013,172,20.doi:10.1016/j.micromeso.2013.01.007
(19)Wu,X.Q.;Zong,R.L.;Zhu,Y.F.Acta Phys.-Chim.Sin.2012,28,437.[吳小琴,宗瑞隆,朱永法.物理化學學報,2012,28,437.]doi:10.3866/PKU.WHXB201112082
(20)Sui,N.;Duan,Y.Z.;Jiao,X.L.;Chen,D.R.J.Phys.Chem.C 2009,113,8560.
(21)Lin,J.;Cai,F.;Zhang,G.Y.;Yang,L.F.;Yang,J.Y.;Fang,W. P.Acta Phys.-Chim.Sin.2013,29,597.[林健,蔡釩,張國玉,楊樂夫,楊金玉,方維平.物理化學學報,2013,29,597.]doi:10.3866/PKU.WHXB201301041
(22)Liang,S.H.;Teng,F.;Bulgan,G.;Zong,R.L.;Zhu,Y.F. J.Phys.Chem.C 2008,112,5307.doi:10.1021/jp0774995
(23)Xu,R.;Wang,X.;Wang,D.S.;Zhou,K.B.;Li,Y.D.J.Catal. 2006,237,426.doi:10.1016/j.jcat.2005.10.026
(24)Dai,Y.;Li,J.H.;Peng,Y.;Tang,X.F.Acta Phys.-Chim.Sin. 2012,28,1771.[戴韻,李俊華,彭悅,唐幸福.物理化學學報,2012,28,1771.]doi:10.3866/PKU.WHXB201204175
(25)Li,J.W.;Song,C.;Liu,S.T.Acta Chim.Sin.2012,70,2347.[李經緯,宋燦,劉善堂.化學學報,2012,70,2347.]doi:10.6023/A12080562
(26)Bastos,S.S.T.;órf?o,J.J.M.;Freitas,M.M.A.;Pereira,M.F. R.;Figueiredo,J.L.Appl.Catal.B:Environ.2009,93,30.doi:10.1016/j.apcatb.2009.09.009
(27)Morales,M.R.;Barbero,B.P.;Cadús,L.E.Appl.Catal.B:Environ.2006,67,229.doi:10.1016/j.apcatb.2006.05.006
(28)Delimaris,D.;Ioannides,T.Appl.Catal.B:Environ.2008,84,303.doi:10.1016/j.apcatb.2008.04.006
(29)Fu,X.B.;Feng,J.Y.;Wang,H.;Ng,K.M.Catal.Commun. 2009,10,1844.doi:10.1016/j.catcom.2009.06.013
(30)Hu,J.;Sun,K.Q.;He,D.P.;Xu,B.Q.Chin.J.Catal.2007,28,1025.[胡敬,孫科強,何代平,徐柏慶.催化學報,2007,28,1025.]doi:10.1016/S1872-2067(08)60001-7
(31)Ye,Q.;Huo,F.F.;Yan,L.N.;Wang,J.;Cheng,S.Y.;Kang,T. F.Acta Phys.-Chim.Sin.2011,27,2872.[葉青,霍飛飛,閆立娜,王娟,程水源,康天放.物理化學學報,2011,27,2872.]doi:10.3866/PKU.WHXB20112872
(32)Dai,Y.;Wang,X.Y.;Li,D.;Dai,Q.G.J.Hazard.Mater.2011,188,132.doi:10.1016/j.jhazmat.2011.01.084
(33)Liu,J.;Makwana,V.;Cai,J.;Suib,S.L.;Aindow,M.J.Phys. Chem.B 2003,107,9185.doi:10.1021/jp0300593
Complete Catalytic Oxidation of Ethanol over MnO2with Different Crystal Phase Structures
ZHANG JieZHANG Jiang-HaoZHANG Chang-BinHE Hong*
(State Key Joint Laboratory of Environmental Simulation and Pollution Control,Center for Air Pollution Control Technology,Research Center for Eco-Environmental Sciences,Chinese Academy of Sciences,Beijing 100085,P.R.China)
α-MnO2,β-MnO2,γ-MnO2,and δ-MnO2catalysts were synthesized by hydrothermal methods,and their catalytic performances towards the oxidation of ethanol were evaluated in detail.The as-synthesized MnO2catalysts were characterized by N2adsorption-desorption measurements,X-ray diffraction(XRD),X-ray photoelectron spectroscopy(XPS),and H2temperature-programmed reduction(H2-TPR).The α-MnO2catalyst showed the best activity of the catalysts tested for the combustion of ethanol and the trend in the activity of different MnO2catalysts towards the oxidation of ethanol was of the order α-MnO2>δ-MnO2>γ-MnO2>β-MnO2. The effect of the crystal phase structure on the activity of the MnO2catalysts was investigated.The XRD results showed that there were differences in the crystallinities of the α-,β-,γ-,δ-MnO2catalysts,but these differences did not have a significant effect on their catalytic performances towards the oxidation of ethanol.The BET surface areas of the α-,β-,γ-,δ-MnO2catalysts exhibited similar tendencies to their ethanol oxidation activities,although the results of standardization calculations showed that the surface area was not the main factor affecting their catalytical activities.The XPS results showed that the lattice oxygen concentration played an important role in defining the catalytic performance of the MnO2.The α-MnO2catalyst showed the best reducibility of all of the MnO2catalysts tested,as determined by H2-TPR.The excellent performance of α-MnO2was attributed to its higher lattice oxygen concentration and reducibility,which were identified as the main factors affecting the activity of the MnO2towards the complete oxidation of ethanol.
November 3,2014;Revised:December 5,2014;Published on Web:December 8,2014.
MnO2catalyst;Low temperature catalytic oxidation;Ethanol complete oxidation;Volatile organic compound;Lattice oxygen
O643
10.3866/PKU.WHXB201412081
The project was supported by the National Natural Science Foundation of China(21422706)and National High Technology Research and Development Program of China(863)(2012AA062702).
國家自然科學基金(21422706)及國家高技術研究發展計劃項目(863)(2012AA062702)資助
?Editorial office ofActa Physico-Chimica Sinica
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
