血流量限制結(jié)合低強(qiáng)度抗阻訓(xùn)練對(duì)下肢骨科術(shù)后肌肉功能的影響
2022-04-25 07:41:04黃凱榮張國(guó)興王俊陳翠珍曾文娣林俊毅李芳鑫
中國(guó)康復(fù) 2022年4期
骨科術(shù)后康復(fù)的主要目標(biāo)是解決患者的疼痛,有效地重建正常的關(guān)節(jié)運(yùn)動(dòng),恢復(fù)肌肉力量和功能
。美國(guó)運(yùn)動(dòng)醫(yī)學(xué)建議傳統(tǒng)肌力訓(xùn)練負(fù)荷需超過(guò)65%~70%最大一次重復(fù)收縮(1 Repetition Maximum, 1RM)才能增加骨骼肌的大小和力量
。為保護(hù)經(jīng)手術(shù)修復(fù)的肢體,手術(shù)后患者常因疼痛、腫脹、關(guān)節(jié)不穩(wěn)或個(gè)人意愿等因素?zé)o法進(jìn)行傳統(tǒng)抗阻肌力訓(xùn)練
。故在較低強(qiáng)度下運(yùn)動(dòng)
,可能導(dǎo)致肌肉力量恢復(fù)受限制
。血流量限制結(jié)合低強(qiáng)度抗阻訓(xùn)練(low load blood flow restriction training,BFRT)是指限制靜脈回流增加肌肉代謝壓力,同時(shí)給予肢體20~30%1RM的阻力進(jìn)行抗阻訓(xùn)練的方法。讓小負(fù)荷低強(qiáng)度的訓(xùn)練也能產(chǎn)生與長(zhǎng)時(shí)間高強(qiáng)度訓(xùn)練相似的生理效果:包括肌肉缺氧酸化、肌肉募集率提高(Ⅱ型肌纖維募集率提高)、神經(jīng)肌肉興奮性增加、生長(zhǎng)激素分泌增加、1型類胰島素生長(zhǎng)因子(insulin-like growth factor-1,IGF-1)和哺乳動(dòng)物雷帕霉素靶蛋白復(fù)合物1:mammalian target of rapamycin complex 1,mTORC1)分泌增多,肌肉生長(zhǎng)抑制素降低
。這種訓(xùn)練方式已經(jīng)從職業(yè)體育領(lǐng)域引用到康復(fù)醫(yī)療領(lǐng)域,越來(lái)越多的文獻(xiàn)證據(jù)表明BFRT具有較高的安全性,在促進(jìn)肌肉和力量的增長(zhǎng)和血管系統(tǒng)健康方面有良好的效果。本研究通過(guò)觀察BFRT對(duì)肌肉功能的影響,為下肢骨科術(shù)后患者提供新的康復(fù)方案。
2.3.3 顏面潮紅發(fā)生率 納入 8 個(gè)研究[6,8,11‐15,17],各研究間為同質(zhì)性(P=0.52,I2=0%),采用固定效應(yīng)模型進(jìn)行Meta‐分析(圖3)。結(jié)果顯示卡貝縮宮素組的顏面潮紅發(fā)生率與縮宮素組無(wú)顯著性差異(OR=0.93,95%CI=0.363~1.36,P=0.70)。
1 資料與方法
1.1 一般資料 本研究開(kāi)始前獲得了廣東省工傷康復(fù)中心倫理委員會(huì)的審查批準(zhǔn),入組前簽署知情同意書。研究對(duì)象為2020年6月~2021年5月在廣東省工傷康復(fù)醫(yī)院住院的下肢骨科術(shù)后患者40例,納入標(biāo)準(zhǔn):膝關(guān)節(jié)及以下組織骨科術(shù)后4~12周;年齡≥16歲,可配合完成治療;排除標(biāo)準(zhǔn):不易控制的高血壓、嚴(yán)重的心肺疾病、血栓性疾病、靜脈曲張、淋巴切除、凝血功能障礙、周圍血管疾病、血管重建等;任何禁止運(yùn)動(dòng)的疾病;不穩(wěn)定性骨折、大腿植皮、傷口未愈及感染者;下肢神經(jīng)損傷,嚴(yán)重的肌肉破壞或壞死;認(rèn)知功能障礙。采用隨機(jī)數(shù)字表法分為觀察組和對(duì)照組,每組20例。其中1例患者因個(gè)人原因退出試驗(yàn),共有39人完成試驗(yàn),觀察組19例,對(duì)照組20例。采用單盲法開(kāi)展研究,即患者對(duì)分組情況不知情。2組患者一般資料比較差異無(wú)統(tǒng)計(jì)學(xué)意義。見(jiàn)表1。

1.2 方法 2組患者均遵循一般術(shù)后康復(fù)訓(xùn)練方案,包括體能耐力訓(xùn)練,關(guān)節(jié)活動(dòng)度訓(xùn)練、負(fù)重訓(xùn)練、本體感覺(jué)訓(xùn)練和步行訓(xùn)練等。訓(xùn)練之前均使用固定式自行車熱身5min,轉(zhuǎn)速為60圈/min。由于患者術(shù)后實(shí)際情況很難獲得真正的1RM,使用病人感覺(jué)為安全的最大負(fù)荷作為1RM。對(duì)照組使用常規(guī)肌力訓(xùn)練方法,根據(jù)患者不同功能情況使用彈力帶、沙袋或醫(yī)學(xué)訓(xùn)練設(shè)備(Medical training therapy,MTT),避免剪切、扭轉(zhuǎn)等不良應(yīng)力刺激。患者以60~80%1RM 作為伸膝抗阻負(fù)荷,完成3組各10次伸膝抗阻訓(xùn)練,組間休息60s。伸膝訓(xùn)練每天1次,每周6d,持續(xù)4周。觀察組使用血流量限制訓(xùn)練帶(blood flow restriction,BFR)進(jìn)行訓(xùn)練,訓(xùn)練前將訓(xùn)練帶內(nèi)空氣排出,環(huán)繞于大腿根部,保證訓(xùn)練帶緊貼皮膚且不滑脫,充氣加壓前無(wú)束縛感,初始?jí)毫x擇200mmHg
。訓(xùn)練帶壓力僅限制靜脈回流,以30%1RM 作為伸膝抗阻負(fù)荷,完成4組抗阻訓(xùn)練,分別是30次,15次,15次,15次,組間休息30s,組間休息保持訓(xùn)練帶壓力直到訓(xùn)練結(jié)束,每次訓(xùn)練時(shí)長(zhǎng)控制小于20min
。根據(jù)個(gè)性化血流限制壓力(Personalized tourniquet pressure,PTP)及訓(xùn)練部位
,在一次訓(xùn)練過(guò)程中不進(jìn)行壓力調(diào)整。每次訓(xùn)練后詢問(wèn)患者疲勞程度,分為4個(gè)等級(jí),分別是無(wú)疲勞、輕度疲勞、疲勞、極度疲勞,下一次訓(xùn)練對(duì)應(yīng)的壓力調(diào)整分別是增加壓力50mmHg、增加壓力25mmHg、保持壓力不變、減低壓力25mmHg
。伸膝訓(xùn)練每天1次,每周6d,持續(xù)4周。
1.3 評(píng)定標(biāo)準(zhǔn) 2組患者均在開(kāi)始訓(xùn)練前,訓(xùn)練4周時(shí)在同一治療師操作下進(jìn)行以下指標(biāo)測(cè)量。①伸膝力量:多關(guān)節(jié)等速肌力評(píng)估系統(tǒng)(BIODEX 4.0,美國(guó))測(cè)量伸膝峰力矩(peak torque)。患者取坐位,配置等速膝關(guān)節(jié)配件,熱身5次,60°/s角速度下完成最大伸膝屈膝向心運(yùn)動(dòng),重復(fù)5次。記錄最大伸膝峰力矩,單位為N·m,代表肌肉收縮的最大肌力。②肌肉形態(tài):肌骨超聲(Terason uSmart 3300)選擇12L5A線陣探頭、肌骨模式膝關(guān)節(jié)部位。患者取臥位,膝下墊滾軸保持屈膝約30°,探頭短軸垂直皮膚表面測(cè)量髕骨上極10cm處股直肌厚度、股直肌橫截面積、股中間肌厚度,使用較多耦合劑并減少加壓,降低對(duì)軟組織的壓迫
。③肢體圍度:患者取臥位伸膝,用軟尺測(cè)量距髕骨上極10cm處測(cè)量大腿肌圍度。④平衡功能:平衡測(cè)試及訓(xùn)練系統(tǒng)(TecnoBody PK252,意大利)測(cè)量睜眼靜態(tài)穩(wěn)定極限,患者按照要求站立在平衡板上,朝8個(gè)方向分別進(jìn)行重心轉(zhuǎn)移,記錄患者完成結(jié)果。
前些日子,老伴突然暈倒,住院不到一個(gè)月就去世了。聽(tīng)醫(yī)生說(shuō),老伴早就有發(fā)病的征兆,只是誰(shuí)都沒(méi)在意,耽誤了治療。三個(gè)女兒為她們疏忽老爸的生活愧疚不已,也對(duì)我平時(shí)對(duì)老伴的不管不問(wèn)很是生氣。
2 結(jié)果
本研究發(fā)現(xiàn)低負(fù)荷運(yùn)動(dòng)中增加BFR訓(xùn)練帶在改善下肢骨科術(shù)后患者肌肉功能具有積極作用,4周治療后患者的伸膝峰力矩、肌肉形態(tài)、肢體圍度、靜態(tài)穩(wěn)定極限等數(shù)據(jù)均有明顯提升。股直肌厚度、股直肌橫截面積、肢體圍度和靜態(tài)穩(wěn)定極限均與對(duì)照組結(jié)果一致,組間差異不明顯。伸膝峰力矩的治療效果觀察組明顯優(yōu)于常規(guī)訓(xùn)練,且僅觀察組患者股中間肌厚度前后變化有統(tǒng)計(jì)學(xué)意義。可能與以下情況有關(guān):第一,術(shù)后患者出現(xiàn)肌肉萎縮、肌力下降是常見(jiàn)的現(xiàn)象,但因疼痛、腫脹、不穩(wěn)或個(gè)人意愿等因素?zé)o法進(jìn)行高強(qiáng)度抗阻肌力訓(xùn)練,延遲肌肉及功能的恢復(fù)。使用BFRT訓(xùn)練帶結(jié)合低負(fù)荷抗阻訓(xùn)練,患者訓(xùn)練時(shí)配合程度高,耐受情況更好。BFRT通過(guò)限制靜脈回流增加肌肉代謝壓力,促進(jìn)生長(zhǎng)激素的分泌和生成,讓小負(fù)荷低強(qiáng)度的訓(xùn)練也能產(chǎn)生與高強(qiáng)度抗阻肌力訓(xùn)練相似的生理效果
。許多研究證明BFRT能有效增加肌力和肌肉大小,Scott 等
通過(guò)研究證明在低負(fù)荷有氧運(yùn)動(dòng)中,BFRT可顯著改善肌肉的形態(tài)和增加力量,并闡述了BFRT促進(jìn)肌肉生長(zhǎng)效果的最佳條件。Pearson 等
闡述了BFRT產(chǎn)生肌肥大效果的工作原理,為BFRT提供了理論依據(jù)。Hughes和Mason等
通過(guò)實(shí)驗(yàn)證明BFRT對(duì)前交叉韌帶重建術(shù)后患者肌肉功能恢復(fù)有促進(jìn)作用。第二,所有患者的一般術(shù)后康復(fù)訓(xùn)練方案,包括體能耐力訓(xùn)練,關(guān)節(jié)活動(dòng)度訓(xùn)練、負(fù)重訓(xùn)練、本體感覺(jué)訓(xùn)練和步行訓(xùn)練等,不能完全控制訓(xùn)練量一致,肌力訓(xùn)練方法也不盡相同,均可能會(huì)對(duì)訓(xùn)練結(jié)果造成一定影響。第三,肌肉厚度是影響肌肉力量的重要因素,許多BFRT使用肌骨超聲對(duì)肌肉進(jìn)行評(píng)估
,但肌肉形態(tài)可能受軟組織水腫、肌肉邊界不清晰、探頭加壓力度等影響。本研究選取術(shù)后4周以后患者進(jìn)行檢測(cè),一方面患者或已出現(xiàn)明顯的肌肉萎縮,另一方面患者的軟組織水腫得到控制,盡可能減小水腫的影響。第四,股四頭肌由股直肌、股中間肌、股外側(cè)肌和股內(nèi)側(cè)肌組成,其中股直肌是連接髖關(guān)節(jié)和膝關(guān)節(jié)的雙關(guān)節(jié)肌肉,股中間肌對(duì)于伸膝爆發(fā)力作用明顯。Cheon
的研究認(rèn)為雖然這組肌肉均起到膝伸肌作用,但每個(gè)肌肉有特定的功能,不同的訓(xùn)練模式對(duì)肌肉刺激效果不同。Ryosuke
認(rèn)為在構(gòu)成股四頭肌的四塊肌肉中,股中間肌的肌肉結(jié)構(gòu)是膝關(guān)節(jié)力量的最佳預(yù)測(cè)指標(biāo)。本研究中觀察組及對(duì)照組的訓(xùn)練負(fù)荷不同,僅觀察組患者股中間肌厚度前后有明顯變化,且治療前后觀察組伸膝峰力矩較對(duì)照組變化明顯可能與此有關(guān)。
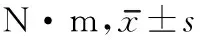
2.3 肢體圍度 治療前2組患者肌圍度未見(jiàn)明顯差異。治療4周后,2組患者肌圍度均較治療前增加(
<0.05)。組間比較無(wú)明顯差異。見(jiàn)表4。
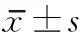
2.2 肌肉形態(tài) 2組患者治療前股直肌厚度、股直肌橫截面積、股中間肌厚度均無(wú)明顯差異。治療4周后,2組患者的股直肌厚度、股直肌橫截面積均較治療前明顯提高(
<0.05),但2組間比較無(wú)明顯差異;觀察組患者股中間肌厚度較治療前有明顯增加(
<0.05),對(duì)照組患者股中間肌厚度治療前后未見(jiàn)明顯差異。見(jiàn)表3。
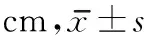
2.4 靜態(tài)穩(wěn)定極限 治療前2組患者靜態(tài)穩(wěn)定極限無(wú)明顯差異。治療4周后,2組患者穩(wěn)定極限較治療前明顯提高(
<0.01)。組間比較無(wú)明顯差異。見(jiàn)表5。

3 討論
2.1 伸膝峰力矩 治療前2組患者伸膝峰力矩差異無(wú)顯著意義。治療4周后,2組患者伸膝峰力矩均較治療前明顯增加(
<0.01),且觀察組優(yōu)于對(duì)照組(
<0.01)。見(jiàn)表2。
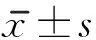
近年來(lái),金融改革持續(xù)深化,民營(yíng)銀行、村鎮(zhèn)銀行等相繼設(shè)立,其成立時(shí)間短、資本規(guī)模小、經(jīng)營(yíng)管理能力和抗風(fēng)險(xiǎn)能力相對(duì)較弱,出現(xiàn)破產(chǎn)的風(fēng)險(xiǎn)相對(duì)較高。同時(shí),我國(guó)銀行業(yè)對(duì)外開(kāi)放的速度、幅度和深度不斷擴(kuò)大。2017年底,外資銀行在華營(yíng)業(yè)性機(jī)構(gòu)總數(shù)已達(dá)1013家。外資入股中資銀行的股比限制被正式取消,中外資適用統(tǒng)一的市場(chǎng)準(zhǔn)入和行政許可辦法。在此背景下,建立適用于包括民營(yíng)銀行、外資銀行在內(nèi)的問(wèn)題銀行市場(chǎng)化退出機(jī)制迫在眉睫。
另外,安全性是BFRT應(yīng)用的前提。Bond等
對(duì)骨科術(shù)后使用BFRT的安全性問(wèn)題進(jìn)行研究,對(duì)比BFR與深靜脈血栓(Deep Vein Thrombosis,DVT)之間關(guān)系,認(rèn)為BFRT不會(huì)導(dǎo)致血流凝滯、血管內(nèi)壁損傷或使血液處于高凝狀態(tài),不會(huì)增加DVT的風(fēng)險(xiǎn)。但是DVT的發(fā)生是相當(dāng)嚴(yán)重的,要基于臨床醫(yī)生對(duì)BFRT生理機(jī)制的理解,以及對(duì)風(fēng)險(xiǎn)的預(yù)測(cè),訓(xùn)練前可使用骨科術(shù)后靜脈血栓風(fēng)險(xiǎn)相關(guān)的BFRT相對(duì)禁忌癥進(jìn)行篩查。
對(duì)于供應(yīng)方,供應(yīng)方不再充當(dāng)被雇傭的角色,而是可以作為主體直接向需求方提供服務(wù),并且獲得相應(yīng)的報(bào)酬。一般租賃或者服務(wù)軟件都要向需求者收取一定的傭金才能提供服務(wù),而在共享經(jīng)濟(jì)下的個(gè)人服務(wù)者無(wú)需支付任何費(fèi)用作為傭金。此外,共享經(jīng)濟(jì)平臺(tái)為供應(yīng)者提供大量的客源,供應(yīng)者在此平臺(tái)上只需要進(jìn)行簡(jiǎn)單的注冊(cè),通過(guò)身份認(rèn)證和服務(wù)考核就可以通過(guò)平臺(tái)尋找客源,減少了因?yàn)檎也坏叫枨笳叨鴮?dǎo)致的資源閑置和浪費(fèi)供應(yīng)者的時(shí)間。最重要的就是共享經(jīng)濟(jì)給供應(yīng)方帶來(lái)的經(jīng)濟(jì)效益,這是每個(gè)供應(yīng)者最關(guān)注的一點(diǎn)。閑置資源是一種浪費(fèi),而共享經(jīng)濟(jì)使閑置資源為供應(yīng)者帶來(lái)效益,同時(shí)也為整個(gè)市場(chǎng)帶來(lái)了更多的有效供給,這在一定程度上促進(jìn)了綠色經(jīng)濟(jì)的發(fā)展。
綜上所述,BFRT可改善患者肌肉功能,增加伸膝峰力矩和股中間肌厚度效果優(yōu)于常規(guī)訓(xùn)練,在骨科康復(fù)領(lǐng)域具有較高的臨床意義。
[1] Bond CW, Hackney KJ, Brown SL, et al. Blood Flow Restriction Resistance Exercise as a Rehabilitation Modality Following Orthopaedic Surgery: A Review of Venous Thromboembolism Risk. J Orthop Sports Phys Ther. 2019,49(1):17-27.
[2] American College of Sports Medicine. American College of Sports Medicine position stand. Progression models in resistance training for healthy adults[J]. Med Sci Sports Exerc. 2009,41(3):687-708.
[3] Hopkins J, Ingersoll C. Arthrogenic muscle inhibition: a limiting factor in joint rehabilitation[J]. J Sport Rehabil, 2000,9(2):135-159.
[4] van Grinsven S, van Cingel RE, Holla CJ, et al. Evidence-based rehabilitation following anterior cruciate ligament reconstruction[J]. Knee Surg Sports Traumatol Arthrosc, 2010,18(8):1128-1144.
[5] Palmieri-Smith RM, Thomas AC, Wojtys EM. Maximizing quadriceps strength after ACL reconstruction[J]. Clin Sports Med, 2008,27(3):405-424.
[6] Vanwye WR, Weatherholt AM, Mikesky AE. Blood Flow Restriction Training: Implementation into Clinical Practice[J]. Int J Exerc Sci, 2017,10(5):649-654.
[7] Ilett MJ, Rantalainen T, Keske MA, et al. The Effects of Restriction Pressures on the Acute Responses to Blood Flow Restriction Exercise[J]. Front Physiol, 2019,10(8):1018.
[8] Bryk FF, Dos Reis AC, Fingerhut D, et al. Exercises with partial vascular occlusion in patients with knee osteoarthritis: a randomized clinical trial[J].?Knee Surg Sports Traumatol Arthrosc. 2016,24(5):1580-1586.
[9] Yasuda T, Fukumura K, Fukuda T, et al. Effects of low-intensity, elastic band resistance exercise combined with blood flow restriction on muscle activation.?Scand J Med Sci Sports[J]. 2014,24(1):55-61.
[10] Bordessa JM, Hearn MC, Reinfeldt AE, et al. Comparison of blood flow restriction devices and their effect on quadriceps muscle activation[J].?Phys Ther Sport. 2021,49(5):90-97.
[11] Lopez P, Pinto MD, Pinto RS. Does Rest Time before Ultrasonography Imaging Affect Quadriceps Femoris Muscle Thickness, Cross-Sectional Area and Echo Intensity Measurements[J].?Ultrasound Med Biol. 2019,45(2):612-616.
[12] Scott BR, Loenneke JP, Slattery KM, et al. Exercise with Blood Flow Restriction: An Updated Evidence-Based Approach for Enhanced Muscular Development[J]. Sports Med, 2015,45(3):313-325.
[13] Pearson SJ, Hussain SR. A Review on the Mechanisms of Blood-Flow Restriction Resistance Training-Induced Muscle Hypertrophy[J]. Sports Med, 2015,45(2):187-200.
[14] Hughes L, Rosenblatt B, Haddad F, et,al. Comparing the Effectiveness of Blood Flow Restriction and Traditional Heavy Load Resistance Training in the Post-Surgery Rehabilitation of Anterior Cruciate Ligament Reconstruction Patients: A UK National Health Service Randomised Controlled Trial[J]. Sports Med, 2019,49(11):1787-1805.
[15] Mason J, Owens J, Brown W, et al. Blood Flow Restriction Training: Current and Future Applications for the Rehabilitation of Musculoskeletal Injuries[J]. Tech Orthop, 2018,33(2):106-113.
[16] Gavanda S, Isenmann E, Schl?der Y, et al. Low-intensity blood flow restriction calf muscle training leads to similar functional and structural adaptations than conventional low-load strength training: A randomized controlled trial[J]. PLoS One, 2020,15(6): e0235377.
[17] Wengle L, Migliorini F, Leroux T, et al. The Effects of Blood Flow Restriction in Patients Undergoing Knee Surgery: A Systematic Review and Meta-analysis [J]. Am J Sports Med, 2021,10(8):3635465211027296.
[18] Cheon S, Lee JH, Jun HP, et al. Acute Effects of Open Kinetic Chain Exercise Versus Those of Closed Kinetic Chain Exercise on Quadriceps Muscle Thickness in Healthy Adults[J]. Int J Environ Res Public Health, 2020,17(13):4669.
[19] Ando R, Saito A, Umemura Y, et al. Local architecture of the vastus intermedius is a better predictor of knee extension force than that of the other quadriceps femoris muscle heads[J]. Clin Physiol Funct Imaging, 2015,35(5):376-382.
