輔料對消瘀康片薄層色譜鑒別的影響
2015-10-19 02:17:32韓曉萍卜晨琛王慧春
中成藥 2015年1期
韓曉萍, 卜晨琛, 王慧春, 海 平
(青海省食品藥品檢驗所,青海 西寧 810016)
輔料對消瘀康片薄層色譜鑒別的影響
韓曉萍,卜晨琛,王慧春,海 平
(青海省食品藥品檢驗所,青海西寧810016)
目的 分別對消瘀康膠囊劑和片劑進行薄層色譜鑒別研究,考察片劑中的工藝輔料對兩制劑中同一藥味薄層色譜鑒別的影響。方法 采用薄層色譜法,分別對兩制劑中木香、蘇木、乳香-沒藥、甘草等藥味進行鑒別試驗。結(jié)果在相同的提取和鑒別條件下,消瘀康膠囊中各檢測藥味的薄層色譜鑒別分離良好,而消瘀康片因工藝中添加了極性輔料造成各藥味斑點拖尾、擴散、分離效果差,為達到滿意效果,比較不加輔料的膠囊,片劑樣品需進行進一步凈化。結(jié)論 工藝輔料對消瘀康片的薄層色譜鑒別存在較為明顯的影響,在標準提高研究工作中,系列品種之間質(zhì)量標準中的鑒別方法不能全盤搬用,而應因品種而異,充分考慮工藝中引入的輔料對薄層色譜鑒別產(chǎn)生的影響,必要的時候應采用合適的方法將其影響去除。
工藝輔料;消瘀康片;薄層色譜鑒別
消瘀康由當歸、川芎、蘇木、木香、沒藥、乳香、赤芍、地黃等17味藥物組方而成,具有活血化瘀,消腫止痛之功效,用于治療顱內(nèi)血腫吸收期,治療腦出血安全有效[1]。國家食品藥品監(jiān)督管理總局網(wǎng)站數(shù)據(jù)查詢顯示批準生產(chǎn)的有消瘀康膠囊和消瘀康片兩個劑型,為不同的廠家生產(chǎn)。筆者在對此品種進行標準提高研究工作中發(fā)現(xiàn),在木香、蘇木、乳香-沒藥、甘草等藥味的薄層色譜鑒別中,在相同的提取和鑒別條件下,消瘀康膠囊中上述藥味的色譜斑點分離良好,主斑點Rf值合適,而消瘀康片的色譜斑點拖尾嚴重、分離欠佳,分析原因是工藝中的輔料造成了這種差異。兩品種的工藝制法表明消瘀康膠囊中未加輔料,僅為乳香、沒藥的細粉和其余藥味的干浸膏制成,而消瘀康片中相應加入了低取代羥丙纖維素、交聯(lián)聚維酮、微晶纖維素、微晶硅膠等輔料,這些極性輔料影響了目標測定物質(zhì)在硅膠板上的吸附,在上行展開時主斑點拖尾、不集中,需對片劑的供試品進行進一步的處理和凈化才能獲得滿意的色譜圖。本研究就首次對中藥制劑中存在的工藝輔料對薄層色譜鑒別的影響進行分析研究。
1 儀器與試藥
METTLER XS105DU電子天平;Sartorius CPA 225D電子天平;Sartorius BSA 224S電子天平;KQ5200V超聲波清洗器;HH-6數(shù)顯恒溫水浴鍋;CAMMA ATS4自動點樣儀,CAMMA reprostor3薄層色譜成像系統(tǒng)。
消瘀康膠囊樣品3批(樣品批號分別為20111102、20120101、20120404,青海益欣藥業(yè)有限責任公司提供,規(guī)格0.4 g/粒);消瘀康片樣品3批(批號20120101、20120102、20120103,由吉林吉爾吉藥業(yè)有限公司提供,規(guī)格0.62 g/片);對照藥材當歸(120927-201014)、川芎(121918-200406)、地黃(121180-200604)、乳香(907-200202)、沒藥(121250-200503)、木香(120921-201008)、蘇木(1067-200202)、甘草(120904-200914)均購自中國食品藥品檢定研究院;硅膠G薄層板(批號20120208,青島海洋化工廠),試劑均為分析純,水為超純水。
2 試驗與方法
2.1木香薄層色譜鑒別[2-5]取消瘀康片10片,除去薄膜衣,研細,稱取2.5 g(膠囊劑稱取1 g),加入甲醇10 mL,超聲處理5 min,濾過,取續(xù)濾液,即為供試品溶液。另外稱取1 g木香對照藥材,同此法制備成木香對照藥材溶液。分別吸取兩種溶液各5μL點至同一硅膠G薄層板上,展開劑為甲苯-甲醇(27∶1),展開,取出,晾干,噴5%香草醛硫酸溶液作為顯色劑,加熱到斑點明顯,日光下檢視,結(jié)果見圖1。
2.2蘇木薄層色譜鑒別[2-5]取消瘀康片15片,除去薄膜衣,研細,稱取5 g(膠囊劑稱取1 g),加入乙醇30 mL,超聲處理5 min,用脫脂棉濾過,取濾液蒸干,殘渣加乙醇2 mL溶解,即為供試品溶液。另稱取1 g蘇木對照藥材,加乙醇10 m L,超聲處理30 min,取續(xù)濾液作蘇木對照藥材溶液。分別吸取上述兩種溶液各5μL點至同一硅膠G薄層板上,展開劑為甲苯-醋酸乙酯-水-甲酸(20∶10∶1∶1)的上層溶液,展開,取出,晾干,在日光下檢視,結(jié)果見圖1。
2.3乳香、沒藥薄層色譜鑒別[6-8]取消瘀康片10片,除去薄膜衣,研細,稱取3 g(膠囊劑稱取1.5 g),加乙醚30 mL,超聲處理20 min,濾過,取濾液,用水洗滌3次,每次30 mL,棄去水洗液,乙醚提取液低溫揮干,殘渣加無水乙醇1 m L溶解作供試品溶液。另取乳香、沒藥對照藥材各0.5 g,分別加乙醚20 mL,超聲處理20 min,濾過,濾液揮干,殘渣加無水乙醇1 mL使溶解,作為乳香、沒藥對照藥材溶液。分別吸取上述供試品溶液10μL、對照藥材溶液5μL點至同一硅膠G薄層板上,以正己烷-乙酸乙酯(14∶4)為展開劑,展開,取出,晾干,噴5%香草醛硫酸溶液作顯色劑,加熱顯色,日光下檢視,結(jié)果見圖1。
2.4甘草薄層色譜鑒別[2-5]取消瘀康片15片,除去薄膜衣,研細,稱取5 g(膠囊劑稱取1 g),加甲醇10 mL,超聲處理5 min,濾過,取續(xù)濾液作為供試品溶液。稱取1 g甘草對照藥材,依上述方法處理,制成甘草對照藥材溶液。分別吸取上述兩種溶液各3μL點至同一硅膠G薄層板上,展開劑為甲苯-醋酸乙酯-水-甲酸(20∶10∶1∶1)上層溶液,展開,取出,晾干,在紫外光燈(365 nm)下檢視,結(jié)果見圖1。

圖1 五味藥材原方法下的薄層色譜圖
3 結(jié)果與分析
3.1上述五味藥材的TLC研究結(jié)果表明,消瘀康膠囊中木香、蘇木、乳香-沒藥、甘草薄層色譜鑒別的色譜斑點分離良好,主斑點Rf值合適;而消瘀康片的色譜斑點拖尾嚴重,分離欠佳。文獻[9-11]表明,藥品輔料能影響到藥物的含量測定、安全性、溶出度等方面,分析本研究中消瘀康片薄層色譜鑒別中存在的問題,認為是片劑工藝中輔料的存在造成了這種差異。兩品種的工藝制法表明消瘀康膠囊僅為乳香、沒藥的細粉和其余藥味的干浸膏制成,未加輔料;而消瘀康片中相應加入了低取代羥丙纖維素、交聯(lián)聚維酮、微晶纖維素、微晶硅膠等極性輔料,這些輔料的存在影響了目標物質(zhì)在硅膠板上的吸附和展開,在上行展開時主斑點拖尾、不集中,需對供試品進行進一步的處理和凈化才能獲得合理的色譜圖。
3.2同時對兩制劑中川芎-當歸和地黃按原標準方法[2-3]進行了薄層色譜鑒別試驗,結(jié)果顯示這幾味藥材的薄層色譜鑒別在膠囊和片劑同樣處理方式和展開系統(tǒng)下均合理可行,分析認為消瘀康片中加入的輔料都是極性大分子物質(zhì),在用非極性溶劑提取,非極性展開系統(tǒng)展開的條件下輔料對斑點展開的影響較小。
4 討論
4.1薄層色譜分離是利用各成分對同一吸附劑的吸附能力不同,使在移動相(溶劑)流過固定相(吸附劑)的過程中,連續(xù)地產(chǎn)生吸附、解吸附、再吸附、再解吸附,從而使各成分互相分離[12]。中藥成分復雜,特別是中藥復方成分更是復雜多樣,不同成分之間相互干擾嚴重[13],難以得到滿意的分離效果,因此樣品前處理對于中藥的薄層色譜鑒別至關重要。
4.2針對上述問題,本研究采用在消瘀康片原鑒別方法的基礎上進行進一步的前處理,具體為在每份供試品溶液制備中加一步過中性氧化鋁柱[14-16]的凈化步驟,以消除這些極性輔料對斑點的影響,其余均按“2試驗與方法”項下操作,結(jié)果見圖2。
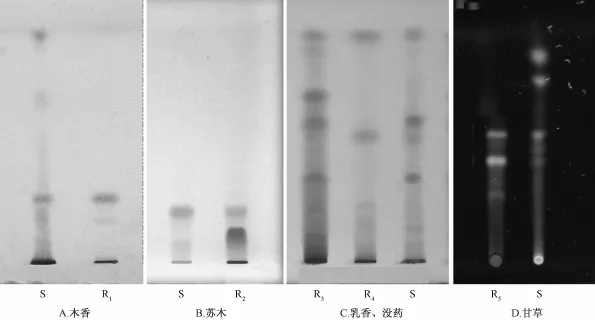
圖2 五味藥材優(yōu)化后的薄層色譜圖
結(jié)果表明經(jīng)過中性氧化鋁柱的凈化處理后,薄層色譜圖譜斑點分離良好,效果較為滿意,說明中性氧化鋁相對硅膠而言競爭性地吸附了供試品溶液中微量的極性輔料,使輔料對色譜斑點的影響減小或消失。
4.3綜上所述,在標準提高研究工作中,系列品種之間的質(zhì)量標準中鑒別方法不能全盤搬用,而因品種而異,充分考慮工藝中引入的輔料對薄層色譜鑒別產(chǎn)生的影響,必要的時候應采用合適的方法將其影響去除。
[1]曹明芳.消瘀康膠囊治療腦出血420例臨床觀察[J].中國醫(yī)藥指南,2012,10(7):225-226.
[2]WS-10766(ZD-0766)-2002,消瘀康膠囊質(zhì)量標準[S].
[3]YBZ05272009,消瘀康片質(zhì)量標準[S].
[4]蔡步林,鄭金鳳,李文莉.回春酒質(zhì)量標準的研究[J].中國藥師,2012,15(9):1241-1243.
[5]梁 云,李彩東,張偉珠.芪復肝顆粒薄層鑒別方法研究[J].中國現(xiàn)代藥物應用,2011,5(20):113-114.
[6]蔣 敏,段海燕.接骨七厘片中血竭、乳香、沒藥的薄層色譜鑒別[J].湖南中醫(yī)學院學報,2000,20(4):30-31.
[7]席桂才,劉麗英.傷痛寧片中乳香的薄層鑒別[J].中華臨床醫(yī)學研究雜志,2008,14(10):1500.
[8]陳 幸,黎萬壽,朱久武.含川牛膝中成藥的薄層鑒別[J].中成藥,1999,21(6):287-289.
[9]尚衛(wèi)平,吳曉芳.安乃近片劑中輔料糊精對主成分含量測定的影響[J].中國藥品標準,2003,4(5):26-27.
[10]張冬梅,宋韶錦,劉 冰.藥用輔料阿司帕坦對格列吡嗪分散片質(zhì)量控制的影響[J].中國藥業(yè),2005,14(2):44.
[11]楊 銳,孫會敏,于麗娜等.藥用輔料對藥品安全性的影響[J].藥物分析雜志,2012,32(7):1309-1314.
[12]李向軍,王 超,王 永,等.中藥薄層色譜影響因素分析及應用[J].中國藥業(yè),2011,20(14):13-15.
[13]謝孟峽,李建新.酚類化合物的反相高效液相色譜研究-溶質(zhì)的溶劑對溶質(zhì)色譜行為的影響[J].北京師范大學學報:自然科學版,1995,31(1):85-90.
[14]徐長金.氧化鋁柱層析-分光光度法測定燒傷Ⅱ號藥中小檗堿的含量[J].時珍國醫(yī)國藥,1998,9(5):423-424.
[15]范文成,葉曉紅,羅志宏.中性氧化鋁柱對絞股藍總皂苷含量測定結(jié)果的影響[J].河北醫(yī)藥,2005,27(2):145.
[16]許海玉,劉 瑩,張鐵軍,等.氧化鋁柱純化吳茱萸提取液中總生物堿和檸檬苦素的研究[J].中草藥,2010,5(41)741-744.
R927.1
B
1001-1528(2015)01-0210-03
10.3969/j.issn.1001-1528.2015.01.047
2013-10-10
韓曉萍(1980—),女,碩士,主管藥師,研究方向:中藥分析及質(zhì)量控制。Te1:13897201414,E-mai1:89323352@qq.com