枯草芽孢桿菌表達系統及其啟動子研究進展
2015-10-26 08:51:16余小霞田健劉曉青伍寧豐
生物技術通報 2015年2期
余小霞 田健 劉曉青 伍寧豐
(中國農業科學院生物技術研究所,北京 100081)
枯草芽孢桿菌表達系統及其啟動子研究進展
余小霞 田健 劉曉青 伍寧豐
(中國農業科學院生物技術研究所,北京 100081)
枯草芽孢桿菌作為一種革蘭氏陽性細菌,由于其具有非致病性、分泌蛋白能力強的特性和良好的發酵基礎及生產技術,是目前原核表達系統中表達和分泌外源蛋白的理想宿主,成為原核表達系統中的一種重要的模式菌株。而實現外源蛋白的高效表達的關鍵因素之一是使用強并可控制的啟動子。目前,枯草芽孢桿菌中常用的啟動子為組成型、誘導物誘導型、時期特異性及自誘導型。詳細介紹枯草芽孢桿菌表達系統以及其常用啟動子的優缺點,并對克隆新的啟動子的方法做了總結,旨為完善枯草表達系統和工業生產外源蛋白奠定基礎。
枯草芽孢桿菌;表達系統;組成型啟動子;誘導型啟動子
枯草芽孢桿菌(Bacillus subtilis)是一種重要的原核表達宿主,具有很高的應用價值,其培養簡單快速,具有較強的分泌蛋白質的能力、非致病性及良好的發酵基礎和生產技術,是目前生產各種工業用酶,如淀粉酶、蛋白酶和脂肪酶等的理想表達宿主,是微生物研究領域中的一種重要模式菌株。但是目前,枯草芽孢桿菌表達系統仍然存在不完善的地方,大部分外源蛋白在其體內的表達仍舊不高,這主要是因為:(1)枯草芽孢桿菌在其對數生長末期表達和分泌大量的蛋白酶,影響外源蛋白的穩定性和產量;(2)質粒的分化和結構不穩定性,有時外源蛋白的組成型表達會影響質粒的穩定性;(3)毒性蛋白在其體內不易表達;(4)有些外源蛋白的表達分泌到培養基中會影響宿主菌的生長;(5)分子遺傳操作與大腸桿菌相比較困難。這些因素限制了枯草桿菌表達系統的發展。枯草芽孢桿菌168菌株的基因組全序列測序工作已于1997年完成[1],但到目前為止,其基因組中尚有近50%[2]的基因的功能仍未闡明。因此目前針對枯草芽孢桿菌中基因及相關元件功能,以及其對內部及外界環境響應的研究仍是針對該菌基礎研究的重點。
外源蛋白在枯草芽孢桿菌中的高效表達是實現其在工業應用上的重要途徑,而使用強并可控制的啟動子是外源蛋白高效表達的關鍵因素之一,決定著工業生產酶的大規模生產。
本文簡單介紹了枯草芽孢桿菌表達系統的特點和表達目的蛋白時使用的載體系統,重點介紹枯了枯草芽孢桿菌表達系統中的重要組成部分——啟動子,并對啟動子的分類和克隆進行了詳細的敘述,最后總結了枯草芽孢桿菌表達系統的現狀及展望。
1 枯草芽孢桿菌表達系統的概述
早在100多年前,人們就開始對枯草芽孢桿菌進行研究,但早期的工作主要針對其分離鑒定、形態觀察及功能鑒定等進行研究。1958年,Spizizen[3]發現枯草芽孢桿菌可以在自然條件下形成感受態,攝入外源DNA,從而建立了枯草芽孢桿菌化學感受態制備方法。此后,隨著DNA重組技術的發明,枯草芽孢桿菌表達系統成為研究熱點。
枯草芽孢桿菌是一種存在著發育過程的原核生物,該菌在細胞的培養過程中具有典型的生長規律,如生長延滯期、對數期、穩定期、衰亡期及芽孢發育期。芽孢桿菌通常是采用更換不同的σ因子的方式來實現細胞中不同基因的特異性表達。枯草芽孢桿菌中的σ因子有多種[4],其中σA、σB、σC、σD、σH、σI、σX、σW與枯草芽孢桿菌營養生長相關,且枯草芽孢桿菌中的σA相當于大腸桿菌σ70,是營養生長最主要的σ因子,占σ因子總量的90%-95%,負責轉錄持家基因和孢子形成早期基因;而σE、σF、σG、σK與孢子形成有關。枯草芽孢桿菌表達系統的另一特點是其分泌系統,由于枯草芽孢桿菌是革蘭氏陽性細菌,僅有一層細胞膜。因此,蛋白質的分泌無需轉位至周質空間(革蘭氏陰性細菌,如大腸桿菌需經過此步驟)而直接通過細胞質轉移到胞外。通過生物信息學的預測,枯草芽孢桿菌中共有294種蛋白質被預測為分泌型蛋白質[5]。根據分泌型蛋白有無典型的信號肽,可以將其分泌系統分為依賴信號肽的分泌途徑和不依賴信號肽的分泌途徑。由于對不依賴信號肽的分泌途徑的研究很少,且與依賴信號肽的分泌途徑不同,因此又將其稱為非典型分泌途徑[6]。
枯草芽孢桿菌中至少存在4種依賴信號肽的分泌途徑[7]:Sec分泌途徑(Sec-SRP cooperation pathway)、Tat分泌途徑(Twin-arginine translocation pathway)、ABC轉運子途徑(ATP-binding cassette transporters)、假菌絲蛋白輸出途徑(Pseudopilin export pathway)。Sec分泌途徑是枯草芽孢桿菌中的主要分泌途徑,經此系統分泌的蛋白質含有Sec型信號肽[8];Tat分泌途徑一般只分泌已形成正確構象的胞內蛋白質,經此系統分泌的蛋白質含有Tat型信號肽[8];ABC轉運子途徑主要用于細菌素等小分子的運輸[9];假菌絲蛋白輸出途徑與枯草芽孢桿菌細胞感受態的形成有關[10]。雖然枯草芽孢桿菌有強的胞外蛋白分泌能力,但較遺憾的是外源蛋白在其體內的表達量不高,主要原因有以下兩種:(1)枯草芽孢桿菌表達系統不成熟;(2)枯草芽孢桿菌野生型菌株本身會產生大量的胞外蛋白酶,容易造成目的蛋白的降解。
枯草芽孢桿菌應用的最為廣泛的宿主菌是枯草168菌株,目前已經知道該菌株可產生8種胞外蛋白酶(其中不包括一些未發現的蛋白酶),利用基因突變的方法失活枯草168菌株基因組上中性蛋白酶、堿性蛋白酶、金屬蛋白酶、胞內蛋白酶、芽孢桿菌肽酶F以及中性蛋白酶B六種蛋白酶基因后,將該菌株命名為WB600,其蛋白酶活性仍保留野生型菌株的0.32%[11]。WB700是在WB600的基礎上將絲蛋白酶基因vpr在內的7個蛋白酶基因全失活的突變株,其蛋白酶活力是野生菌株的0.1%。隨后,來自加拿大卡加利大學的Wong等[12]在WB700的基礎上,繼續將枯草桿菌的胞壁蛋白酶基因cwp失活,發展了在野生菌株的基礎上缺失了8個蛋白酶基因的胞外蛋白酶活性更低的WB800菌株。因此,現在用于外源表達目的蛋白常用的枯草桿菌菌株大部分使用的是枯草168系列的突變株。
2 枯草芽孢桿菌表達載體系統
克隆載體和表達載體是基因重組技術的重要工具,在分子遺傳操作的所有領域中處于十分重要的地位。根據類型的不同可將其分為3類[13]:獨立自主復制的質粒載體、整合型載體及噬菌體載體。下面主要介紹在枯草芽孢桿菌中常用的獨立自主復制質粒載體和整合型載體。
2.1 獨立自主復制的質粒載體
枯草芽孢桿菌168系列菌株目前已全部完成測序,成為研究枯草芽孢桿菌的標準菌株,大部分的枯草芽孢桿菌不含有內源性質粒,如今常用的質粒大部分來自于葡萄球菌和鏈球菌[14,15]。這些質粒載體可在枯草芽孢桿菌中自主復制,根據其復制方式可分為滾環復制型載體(Rolling circle-type replication vectors)和θ復制型載體,大部分來自于革蘭氏陽性細菌的小的質粒載體(<12 kb)是通過滾環復制機制復制的,而一些大的質粒載體是通過θ復制機制復制的。如最開始鑒定的來自于金黃色葡萄球菌的4個質粒 pUB110,pC194,pE194和pT181都是通過滾環機制復制的。pUB110帶有卡那霉素的抗性基因,拷貝數較低,每個細胞中有30-50個拷貝[16];pC194帶有氯霉素的抗性基因,拷貝數約為15[17];pE194較特殊,是個溫度敏感性的質粒載體,32℃正常復制,隨著溫度增加,拷貝數降低,45℃時停止復制,這為人們純化含有此類質粒的細胞提供了一個思路。另一方面,pE194可以在細菌基因組上多個位點發生特異性整合,整合子可以通過抗性(紅霉素)和培養溫度(50℃)進行篩選[18,19]。pT181帶有四環素的抗性基因,每個細胞中約有20個拷貝[18]。
這些質粒一般可以在枯草芽孢桿菌中自主復制,表達其抗性基因,但在傳代培養中仍存在許多問題,其中最大的問題是質粒的分離不穩定性和結構不穩定性[20]。質粒分離不穩定性表現在細胞在無篩選壓力條件質粒易丟失;結構不穩定包括質粒DNA的重排、篩減和增加。質粒載體的不穩定性嚴重影響了其在枯草芽孢桿菌的研究應用,如許多基因不能直接構建在枯草芽孢桿菌質粒載體,轉入宿主菌進行表達。然而,大腸桿菌分子遺傳操作成熟,不會產生質粒不穩定性現象,所以構建大腸桿菌-枯草芽孢桿菌的穿梭質粒載體是十分必要的,這樣就可以將外源基因在大腸桿菌中構建完成后再轉化到枯草芽孢桿菌中表達。目前常用的穿梭質粒載體有 pEB10[21]、pEB20[22]、pEB60[22]、pUB18[23]、pUB19[24]和pWB980[24]等,詳細信息,見表1。

表1 常用的大腸桿菌-枯草芽孢桿菌穿梭質粒載體
2.2 整合型質粒載體
芽孢桿菌中高效表達外源蛋白的最大障礙是構建的重組表達質粒在宿主菌中通常不穩定,解決這一問題的有效途徑是使用整合型載體。枯草整合型質粒載體一般含有大腸桿菌質粒的復制起點(通常為pBR322或其衍生質粒)、抗性篩選標記基因(常用于枯草的標記基因有卡那霉素、氯霉素、紅霉素等)及一段或兩段整合基因(即與宿主細胞染色體序列同源的基因)。它的特點是限制性復制,即整合型質粒只在大腸桿菌中有復制功能,轉入革蘭氏陽性細菌如枯草芽孢桿菌中則無此功能,因此只有整合進宿主基因組中才能正常的表達目的基因。整合型質粒載體根據其整合方式不同可分為單交換整合和雙交換整合。
2.2.1 單交換整合 通過一段位于外源基因某側的同源序列(與枯草芽孢桿菌基因組同源的序列),與枯草芽孢桿菌基因組的目標序列發生同源重組,從而將外源基因整合到染色體上,單交換需要的同源臂較短,整合效率高且穩定,不易發生逆整合(圖1)。

圖1 單交換整合型載體
2.2.2 雙交換整合 通過位于外源基因兩側的同源序列,與枯草芽孢桿菌基因組的目標序列發生同源重組。為了提高雙交換的整合效率,其需要的同源臂長度至少要400-500 bp[25],才能發生有效的重組;其次還需要考慮發生重組的線性雙鏈DNA的穩定性。因為線性雙鏈DNA導入枯草芽孢桿菌體內容易被AddAB解旋酶降解,但若在線性雙鏈DNA末端加上Chi位點(χBs,5'-AGCGG-3')可以解決此問題[26,27],因此在構建重組質粒載體時可以在克隆同源臂時引入Chi位點以提高重組效率。目前常用的雙交換整合型載體是通過amyE基因位點進行整合[28],插入的外源基因破壞了amyE基因,宿主菌不能表達淀粉酶,從而可以通過功能平板和酶活測定來鑒定陽性重組子。另外,一些其他的整合型載體還可通過thrC位點進行整合,插入的外源基因破壞thrC基因,從而導致蘇氨酸營養缺陷型[29],以此來篩選重組子(圖2)。常見的整合型載體,見表2。
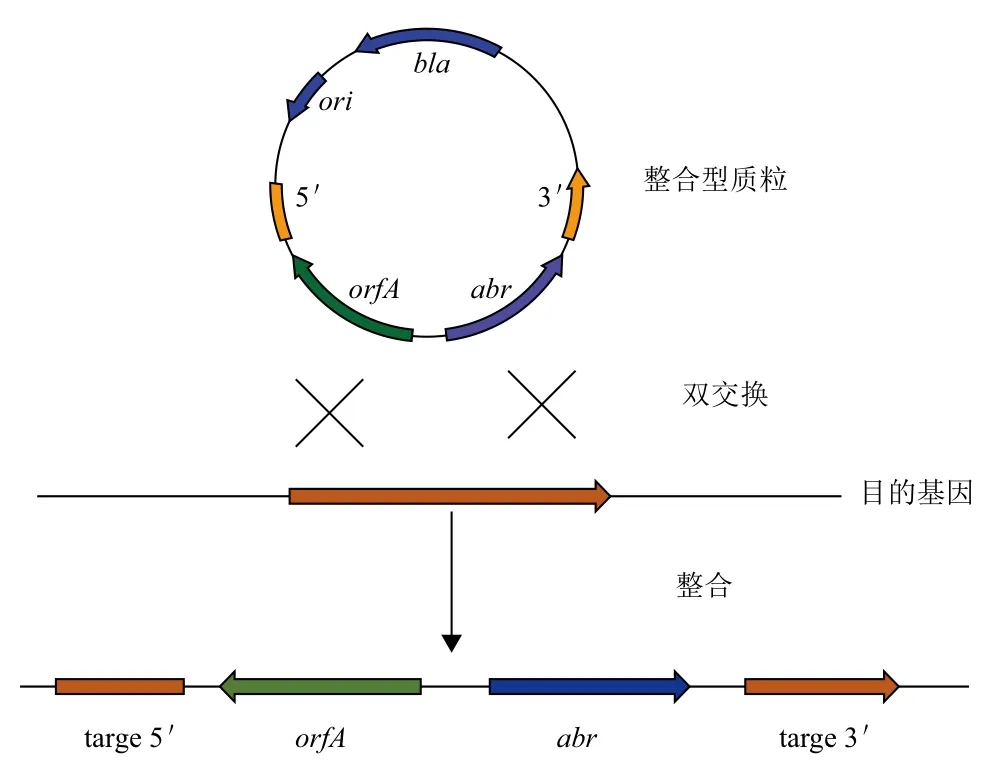
圖2 雙交換整合型載體
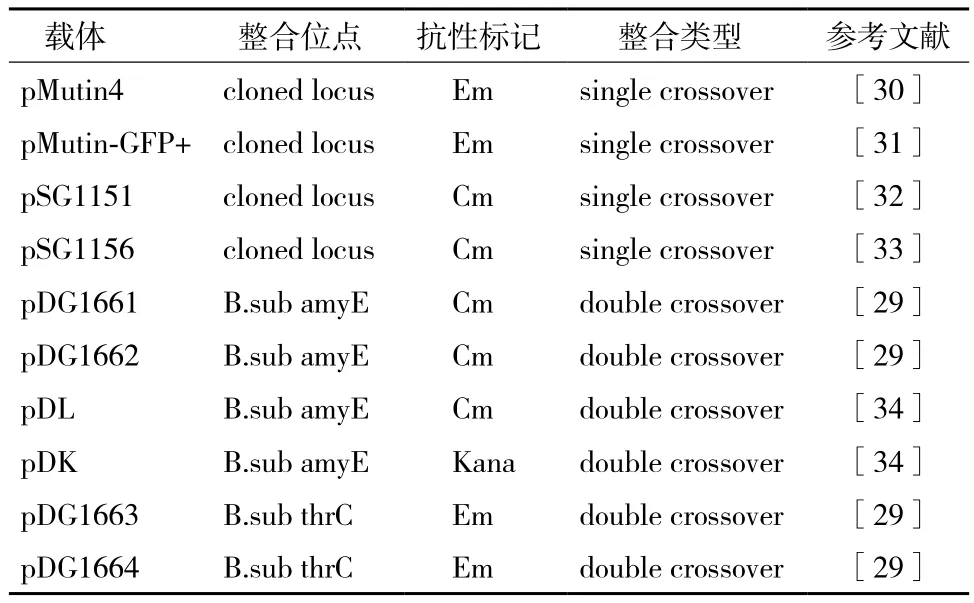
表2 常見的芽孢桿菌整合型載體
3 枯草表達系統的啟動子
啟動子是位于結構基因5'端上游的DNA序列,能活化RNA聚合酶,使之與模板DNA準確的結合并具有轉錄起始的特異性。啟動子是實現外源基因高效表達的關鍵。
3.1 啟動子的分類
依照控制轉錄水平的高低,啟動子可以分為強啟動子和弱啟動子;從啟動子誘導機制上分類,啟動子可以分為組成型啟動子、誘導型啟動子、時期特異性啟動子及自誘導啟動子,下面著重介紹枯草芽孢桿菌中已發表的組成型啟動子和誘導型啟動子[2],并對時期特異性啟動子和自誘導啟動子做簡單的介紹。
3.1.1 組成型啟動子 組成型啟動子就是不需要任何誘導物,可以持續性表達目的蛋白的啟動子。這類啟動子啟動強度通常要求較強,且成本低,一些組成型啟動子常通過構建胞內或分泌表達載體表達目的蛋白。常用的枯草桿菌組成型啟動子有P43啟動子和噬菌體啟動子,其中P43啟動子是枯草桿芽孢菌胞苷脫氨酶(cdd)基因的啟動子,廣泛用于研究,該啟動子屬于重疊啟動子,分別由σA和σB識別,前者負責轉錄持家基因和孢子形成早期基因,后者一般負責與脅迫相關的基因[35]。研究發現P43啟動子為強啟動子,啟動強度強于誘導型啟動子PsacB和PamyE,普遍作為表達載體的調控元件[36]。Yang等[37]通過啟動子誘捕系統(Promoter trap system)從地衣芽孢桿菌基因組中篩選到一個強啟動子Pshuttle-09,其強度是P43的8倍,在分析Pshuttle-09序列的基礎上,Yang等又構建了一個人工雙啟動子Plaps,此啟動子的強度是P43的13倍,是目前應用在枯草芽孢桿菌中表達的最強的組成型啟動子。不過組成型啟動子也存在一些問題,如組成型表達載體不穩定,表達量不易控制,不適合表達對宿主菌有害的或有毒性蛋白,另外重復使用同一種啟動子表達多個外源基因可能會引起基因沉默或共抑制現象等。
3.1.2 誘導型啟動子 與組成型啟動子相比,誘導型啟動子可根據需要在特定的誘導條件,快速誘導基因轉錄的“開”與“關”。誘導型啟動子可以分為兩類:一類與環境應答有關,如滲透壓、pH值、氧氣匱乏、低溫、熱休克等,在這些環境下,啟動子啟動相應基因轉錄,以適應環境變化;另一類是化學誘導啟動子,即受化學誘導物誘導而提高相應基因的表達量。下面主要介紹枯草芽孢桿菌中常用的幾個化學物誘導啟動子。
枯草芽孢桿菌中最常見的是受IPTG誘導的啟動子,IPTG能夠與大腸桿菌的轉錄阻遏蛋白LacI結合,使得LacI從操縱位點上脫離,從而誘導下游基因的表達。最早應用的是啟動子Pspac,由枯草芽孢桿菌噬菌體SPO1和大腸桿菌lac操縱子融合而成[38]。隨后Phan等[39]將熱激蛋白GroES和GroEL編碼序列的強啟動子與lac操縱子融合,構成受IPTG誘導的啟動子Pgrac。Phan等[40]進一步通過優化啟動子Pgrac的調控元件,構建另一個更強的啟動子Pgrac100。雖然受IPTG誘導的啟動子強并且可控,但它存在以下3個缺點:(1)IPTG價格昂貴,且有毒性,不適宜進行大規模的外源蛋白的發酵生產;(2)Pspac啟動強度仍然不夠,外源蛋白的表達量不是很高;(3)Pspac不屬于嚴緊的可控啟動子,從而導致在未加入誘導物IPTG的情況下,仍然有少量的重組蛋白表達。因而這些由IPTG誘導的啟動子不適用于工業上大規模蛋白酶的生產。
枯草芽孢桿菌中第2個啟動子系統是以木糖為誘導物的啟動子Pxyl。Pxyl由xylR編碼的阻遏蛋白嚴格控制表達,通過添加0.1%-2%的木糖,能夠誘導該啟動子的表達,木糖啟動子系統對代謝中的大部分代謝產物不敏感,不過卻受到葡萄糖的抑制[41]。
枯草芽孢桿菌中第3個啟動子系統是以蔗糖為誘導物的sacB啟動子,sacB基因是枯草芽孢桿菌基因組上受蔗糖誘導調控的基因,編碼外分泌的蔗糖果聚糖酶(Levansucrase)。其啟動子sacR轉錄起始是持續性的,不受蔗糖誘導的控制,但在無蔗糖的情況下,其表達強度比有蔗糖誘導時低100倍[42]。
這3個啟動子誘導系統在枯草芽孢桿菌中應用廣泛,尤其是前兩個系統,常被用做外源蛋白高效表達的元件,但它們共同存在一個缺點就是所用的誘導物IPTG和木糖價錢昂貴,在工業生產中易增加成本,因此限制了在工業生產中的應用。而蔗糖誘導啟動子的啟動強度相對較弱,無法在工業生產中高效表達目標蛋白。
除了上述介紹的3種誘導啟動子外,枯草芽孢桿菌中還有受淀粉誘導的持續性表達的淀粉酶基因啟動子Pamy[36],以及受磷酸[43]、檸檬酸鹽[44]、四環素[45]、枯草菌素[46]、細胞壁抗生素[47]和甘氨酸[48]等誘導的啟動子。此外,為了降低誘導物的成本以更利于工業化應用,Heravi等[49]開發了以甘露醇為誘導物的表達系統。Yang等[14,50]利用麥芽糖為誘導物的啟動子Pglv構建了表達系統。Reza等[51]通過施加環境壓力和葡萄糖匱乏構建了PohrB表達系統。
3.1.3 時期特異性啟動子 轉錄組學分析顯示基因表達是有時期依賴性的[52]。一些基因特異性的在對數生長期表達,而另外一些基因卻在菌體進入穩定期后才被激活表達。應用這些時期特異性表達基因的調控元件來表達外源基因,可以避免添加外源的誘導物。目前有兩個此類啟動子被用于外源基因的表達。一個是啟動子PrpsF,特異性在對數期表達,利用此啟動子已成功地將來源于C. perfringens的β-類毒素的基因進行了高效表達[53]。另一個使用的啟動子是在對數末期表達的PaprE。PaprE是σA依賴型的啟動子,且它的活性被高度控制。使用lacZ作為報告基因,將其與啟動子共同構建在枯草桿菌整合型載體上,整合進枯草桿菌基因組進行外源蛋白的表達,結果發現β-半乳糖苷酶的表達量約占總蛋白量的10%[54]。
3.1.4 自誘導啟動子 自誘導啟動子在工業生產中具有優勢。Lee等[55]運用蘇云金芽胞桿菌伴孢晶體蛋白基因cry3Aa的啟動子Pcry3Aa構建了枯草芽孢桿菌穩定期自誘導表達系統,并且通過對此啟動子進行優化,優化后外源蛋白lacZ的表達量較優化前提高了5倍。優化后的Pcry3Aa啟動子可能會成為枯草芽孢桿菌表達系統的一個強的備選啟動子。另外,Wenzel等[56]利用啟動子PmanP構建了適合高細胞密度發酵的自誘導系統。這種新型的枯草芽孢桿菌自誘導系統在工業生產上將不僅可以提高外源蛋白的產量,還可以降低生產成本。
綜上所述,枯草芽孢桿菌表達系統常用的啟動子,見表3。

表3 枯草芽孢桿菌表達系統常用的啟動子
3.2 啟動子克隆的方法
啟動子的研究一方面是為了得到有利于實際應用的高強度啟動子;另一方面也有助于進一步了解基因組和遺傳特性。目前克隆啟動子的方法多種多樣,大致可分為兩種:一是用啟動子誘捕系統(即用到啟動子探針質粒載體)篩選啟動子;另一種根據基因組信息結合計算機軟件預測直接用PCR擴增已知的基因啟動子,然后再進行后期生物實驗驗證,這種方法精確性較差。因此,對于篩選到的啟動子還需要進行進一步的優化。
3.2.1 利用啟動子誘捕系統篩選啟動子 利用啟動子誘捕系統篩選啟動子是一種簡單有效且更加準確的方法[37,57]。啟動子誘捕系統中最為核心的部分是構建啟動子探針質粒載體,這種載體通常由不含啟動子成分的報告基因和一個篩選標記基因組成,只有當內源基因編碼區插入到載體上,序列中產生融合轉錄本時報告基因才可能表達。在探針載體構建時,報告基因的選擇至關重要。作為報告基因,它的全序列必須是已知的、已克隆研究過,另外報告基因的表達產物在宿主菌中不存在背景干擾,并且其表達產物的檢測應簡單、快捷、靈敏度高、重復性好。目前常用的報告基因有以下幾種:(1)β-半乳糖苷酶基因:細菌β-半乳糖苷酶基因,尤其是來自于嗜熱芽孢桿菌的耐熱性的β-半乳糖苷酶基因被廣泛應用,它能水解底物X-gal,利用藍白斑篩選可鑒定插入啟動子的陽性克隆。Yang等[37]利用來自于嗜熱芽孢桿菌的bgaB基因為報告基因構建啟動子捕獲載體pShuttleF,從地衣芽孢桿菌的基因組中篩選出比P43表達強度強8倍的PShuttle-09啟動子;(2)氯霉素轉乙酰酶報告基因(CAT) :細菌CAT酶可以將乙酰基從乙酰輔酶A轉移到氯霉素的3-羥基位上,而使宿主菌失去抗性,再通過檢測放射性標記的底物實現量化。以CAT為報告基因檢測簡單,重現性好且靈敏度高。Harwood等[58]以氯霉素抗性基因為報告基因構建啟動子捕獲載體,在枯草芽孢桿菌中克隆了一些啟動子片段。(3)綠色熒光蛋白報告基因(gfp) :綠色熒光蛋白基因最初是從維多利亞發光水母(Aequorea victoria)中分離得到,在508 nm處自行發射綠色熒光,無需輔助因子和底物。GFP相對分子質量小、對活細胞無毒害、熒光穩定且檢測方法簡單直觀(通過熒光顯微鏡或流式細胞儀檢測),加熱、變性劑及一般的蛋白酶均不能使GFP失活。GFP上述的優點使其成為報告基因的后起之秀。目前已經有多種GFP突變體,如藍色熒光蛋白(CFP)、黃色熒光蛋白(YFP)及加強型的熒光蛋白(EGFP)等。Gat等[57]以gfp和egfp基因為報告基因,通過啟動子誘捕載體從炭疽芽孢桿菌(Bacillus anthracis)中篩選出用于重組蛋白rPA表達的啟動子。此外,還有β-葡萄糖苷酸酶(β-glucuronidase)基因、酸性磷酸脂酶基因及熒光素酶基因等,在啟動子篩選中也應用廣泛。
3.2.2 結合生物信息學和PCR技術克隆啟動子 應用啟動子誘捕載體克隆啟動子無需知道基因的具體序列,可隨機篩選啟動子,這樣不用設計引物,可得到大量的啟動子片段。但這種方法需要構建啟動子文庫,篩選工作量大。隨著生物信息學和PCR技術的發展,利用計算機軟件預測加上PCR擴增和后期的實驗驗證,從基因組中分離已知序列的啟動子逐漸成為熱門。
Wang等[59]在蘇云金芽胞桿菌的RNA測序結果的基礎上,運用生物信息學預測未知基因的轉錄起始位點,并將轉錄起始位點上游500 bp的核苷酸進行分析,尋找與之匹配的σ因子和轉錄因子的識別位點,從而判斷是否存在潛在的可開發的啟動子。使用PCR技術將選定的啟動子克隆并構建到表達載體上,后期進一步通過實驗驗證這些選定的啟動子強度。在此基礎上,他們篩選出20個不同時期表達的啟動子,為蘇云金芽胞桿菌的分子生物學的基礎研究及應用研究奠定了基石。
除了上述的普通PCR擴增啟動子外,還有常用反向PCR、銜接頭PCR及TAIL-PCR等方法擴增啟動子序列。
4 展望
目前,已有許多的原核基因和真核基因在枯草芽孢桿菌中表達。已克隆的原核基因很多,如屬于分子遺傳學范疇的一些與質粒構建有關的抗性基因;與DNA結構有關的DNA重組、誘變、修復等相關基因;與基因表達及其調控有關的Sigma因子、Trna、正負調控基因等;與芽孢形成萌發的spoOA、spoIIG基因等;以及與蘇云金芽胞桿菌伴胞晶體蛋白相關的cry、cyt基因等;甚至有些與感受態、蛋白分泌有關的Com、SecDF基因等。此外,還有一些工業生產用酶也成功在枯草芽孢桿菌中表達,如堿性蛋白酶、中性蛋白酶A和B、α-淀粉酶、β-淀粉酶、支鏈淀粉酶、木聚糖酶、脂肪酶、β-半乳糖苷酶和真枯草多肽酶F等。
真核基因在原核生物中表達總是不理想,其主要原因一是原核生物中缺乏蛋白翻譯后修飾如糖基化等問題;二是枯草桿菌的胞外蛋白酶易降解真核基因產物。因此,其缺失蛋白酶基因的枯草芽孢桿菌成為高表達外源蛋白的理想菌株。
基因表達體系是一個復雜的過程,與大腸桿菌表達系統比較,枯草表達系統還不是很成熟。近年來,科研工作者在枯草芽孢桿菌分泌表達外源蛋白方面取得很大的進展,已建立一個有效的外源蛋白枯草表達系統。相比于傳統的大腸桿菌系統,枯草表達系統要遠遠落后于它,但在蛋白表達純化和分泌活性蛋白方面則要明顯優于大腸桿菌。實踐證明,枯草表達系統可以表達多種可溶性的并具有生物活性的蛋白質。在進一步發展和完善枯草表達系統過程中,勢必要不斷完善和擴大枯草芽孢桿菌載體系統和宿主系統,而在載體系統的改造過程中,啟動子調控元件起著重中之重的作用。因此,從啟動子出發,篩選新的強啟動子元件,構建成熟高效的枯草表達系統,無論是在理論研究還是實際生產中都有十分重要的意義。
[1]Harwood CR, Wipat A. Sequencing and functional analysis of thegenome of Bacillus subtilis strain 168 [J]. FEBS Lett, 1996, 389(1):84-87.
[2]張曉舟. 枯草桿菌新型表達系統和遺傳操作體系的建立及應用[D]. 南京:南京農業大學, 2006.
[3]Spizizen J. Transformation of biochemically deficient strains of Bacillus subtilis by deoxyribonucleate [J]. Proceedings of the National Academy of Sciences of the United States of America, 1958,44(10):1072.
[4]Haldenwang WG. The sigma factors of Bacillus subtilis [J]. Microbiological Reviews, 1995, 59(1):1-30.
[5]Tjalsma H, Bolhuis A, Jongbloed JD, et al. Signal peptide-dependent protein transport in Bacillus subtilis:a genome-based survey of the secretome [J]. Microbiology and Molecular Biology Reviews, 2000,64(3):515-547.
[6]Bendtsen JD, Kiemer L, Fausb?ll A, et al. Non-classical protein secretion in bacteria [J]. BMC Microbiology, 2005, 5(1):58.
[7]Tjalsma H, Antelmann H, Jongbloed JD, et al. Proteomics of protein secretion by Bacillus subtilis:separating the “secrets” of the secretome [J]. Microbiology and Molecular Biology Reviews, 2004,68(2):207-233.
[8]Fu LL, Xu ZR, Li WF, et al. Protein secretion pathways in Bacillus subtilis:Implication for optimization of heterologous protein secretion [J]. Biotechnology Advances, 2007, 25(1):1-12.
[9]Banerjee S, Hansen JN. Structure and expression of a gene encoding the precursor of subtilin, a small protein antibiotic [J]. Journal of Biological Chemistry, 1988, 263(19):9508-9514.
[10]Chung Y, Breidt F, Dubnau D. Cell surface localization and processing of the ComG proteins, required for DNA binding during transformation of Bacillus subtilis [J]. Molecular Microbiology,1998, 29(3):905-913.
[11]Wu XC, Lee W, Tran L, et al. Engineering a Bacillus subtilis expression-secretion system with a strain deficient in six extracellular proteases [J]. Journal of Bacteriology, 1991, 173(16):4952-4958.
[12] Murashima K, Chen CL, Kosugi A, et al. Heterologous production of Clostridium cellulovorans engB, using protease-deficient Bacillus subtilis, and preparation of active recombinant cellulosomes[J]. Journal of Bacteriology, 2002, 184(1):76-81.
[13] Plasmids SB, Harwood CR, Cutting SM(eds)//Molecular biological methods for Bacillus [J]. New York:John Wiley and Sons, 1990:75-174.
[14] Yang MM, Zhang WW, Zhang XF, et al. Construction and characterization of a novel maltose inducible expression vector in Bacillus subtilis [J]. Biotechnology Letters, 2006, 28(21):1713-1718.
[15] Kunst F, Ogasawara N, Moszer I, et al. The complete genome sequence of the gram-positive bacterium Bacillus subtilis [J]. Nature, 1997, 390(6657):249-256.
[16] Lacey R, Chopra I. Genetic studies of a multi-resistant strain of Staphylococcus aureus [J]. Journal of Medical Microbiology, 1974,7(2):285-297.
[17]Iordanescu S, Surdeanu M, Della-Latta P, et al. Incompatibility and molecular relationships between small staphylococcal plasmids carrying the same resistance marker [J]. Plasmid, 1978, 1(4):468-479.
[18]Iord?nescu S. Three distinct plasmids originating in the same Staphylococcus aureus strain [J]. Archives Roumaines de Pathologie Expérimentales et de Microbiologie, 1976, 35(1-2):111.
[19]Hofemeister J, Israeli-Reches M, Dubnau D. Integration of plasmid pE194 at multiple sites on the Bacillus subtilis chromosome [J]. Molecular and General Genetics MGG, 1983, 189(1):58-68.
[20]Bron S, Meijer W, Holsappel S, et al. Plasmid instability and molecular cloning in Bacillus subtilis [J]. Research in Microbiology, 1991, 142(7):875-883.
[21]Bron S, Luxen E, Swart P. Instability of recombinant pUB110 plasmids in Bacillus subtilis:plasmid-encoded stability function and effects of DNA inserts [J]. Plasmid, 1988, 19(3):231-241.
[22]Nagarajan V, Albertson H, Chen M, et al. Modular expression and secretion vectors for Bacillus subtilis [J]. Gene, 1992, 114(1):121-126.
[23]Wang LF, Wong SL, Lee SG, et al. Expression and secretion of human atrial natriuretic α-factor in Bacillus subtilis using the subtilisin signal peptide [J]. Gene, 1988, 69(1):39-47.
[24]Wu SC, Wong SL. Development of improved pUB110-based vectors for expression and secretion studies in Bacillus subtilis [J]. Journal of Biotechnology, 1999, 72(3):185-195.
[25]Melnikov A, Youngman PJ. Random mutagenesis by recombinational capture of PCR products in Bacillus subtilis and Acinetobactercalcoaceticus [J]. Nucleic Acids Research, 1999, 27(4):1056-1062.
[26]Chédin F, Noirot P, Biaudet V, et al. A five-nucleotide sequence protects DNA from exonucleolytic degradation by AddAB, the RecBCD analogue of Bacillus subtilis [J]. Molecular Microbiology,1998, 29(6):1369-1377.
[27]Chedin F, Ehrlich SD, Kowalczykowski SC. The Bacillus subtilis AddAB helicase/nuclease is regulated by its cognate Chi sequence in vitro [J]. Journal of Molecular Biology, 2000, 298(1):7-20.
[28]Hidenori S, Henner DJ. Construction of a single-copy integration vector and its use in analysis of regulation of the trp operon of Bacillus subtilis [J]. Gene, 1986, 43(1):85-94.
[29]Guérout-Fleury AM, Frandsen N, Stragier P. Plasmids for ectopic integration in Bacillus subtilis [J]. Gene, 1996, 180(1):57-61.
[30]Vagner V, Dervyn E, Ehrlich SD. A vector for systematic gene inactivation in Bacillus subtilis [J]. Microbiology, 1998, 144(11):3097-3104.
[31]Kaltwasser M, Wiegert T, Schumann W. Construction and application of epitope-and green fluorescent protein-tagging integration vectors for Bacillus subtilis [J]. Applied and Environmental Microbiology, 2002, 68(5):2624-2628.
[32]Feucht A, Lewis PJ. Improved plasmid vectors for the production of multiple fluorescent protein fusions in Bacillus subtilis [J]. Gene,2001, 264(2):289-297.
[33]Lewis PJ, Marston AL. GFP vectors for controlled expression and dual labelling of protein fusions in Bacillus subtilis [J]. Gene,1999, 227(1):101-109.
[34]Yuan G, Wong SL. Regulation of groE expression in Bacillus subtilis:the involvement of the sigma A-like promoter and the roles of the inverted repeat sequence(CIRCE) [J]. Journal of Bacteriology, 1995, 177(19):5427-5433.
[35]Zhang XZ, Cui ZL, Hong Q, et al. High-level expression and secretion of methyl parathion hydrolase in Bacillus subtilis WB800[J]. Applied and Environmental Microbiology, 2005, 71(7):4101-4103.
[36]Ye RQ, Kim JH, Kim BG, et al. High-level secretory production of intact, biologically active staphylokinase from Bacillus subtilis [J]. Biotechnology and Bioengineering, 1999, 62(1):87-96.
[37]Yang MM, Zhang W, Ji SY, et al. Generation of an artificial double promoter for protein expression in Bacillus subtilis through a promoter trap system [J]. PLoS One, 2013, 8(2):9.
[38]Yansura DG, Henner DJ. Use of the Escherichia coli lac repressor and operator to control gene expression in Bacillus subtilis [J]. Proceedings of the National Academy of Sciences, 1984, 81(2):439-443.
[39]Phan TTP, Nguyen HD, Schumann W. Novel plasmid-based expression vectors for intra-and extracellular production of recombinant proteins in Bacillus subtilis [J]. Protein Expression and Purification, 2006, 46(2):189-195.
[40]Phan TTP, Nguyen HD, Schumann W. Development of a strong intracellular expression system for Bacillus subtilis by optimizing promoter elements [J]. Journal of Biotechnology, 2012, 157(1):167-172.
[41]Kim L, Mogk A, Schumann W. A xylose-inducible Bacillus subtilis integration vector and its application [J]. Gene, 1996, 181(1):71-76.
[42]Zukowski MM, Miller L. Hyperproduction of an intracellular heterologous protein in a sacU sup hsup mutant of Bacillus subtilis[J]. Gene, 1986, 46(2):247-255.
[43]Lee JK, Edwards CW, Hulett FM. Bacillus licheniformis APase I gene promoter:a strong well-regulated promoter in B. subtilis [J]. Journal of General Microbiology, 1991, 137(5):1127-1133.
[44]Yamamoto H, Murata M, Sekiguchi J. The CitST two-component system regulates the expression of the Mg-citrate transporter in Bacillus subtilis [J]. Molecular Microbiology, 2000, 37(4):898-912.
[45]Geissend?rfer M, Hillen W. Regulated expression of heterologous genes in Bacillus subtilis using the Tn10 encodedtet regulatory elements [J]. Applied Microbiology and Biotechnology, 1990, 33(6):657-663.
[46]Bongers RS, Veening JW, Van Wieringen M, et al. Development and characterization of a subtilin-regulated expression system in Bacillus subtilis:strict control of gene expression by addition of subtilin [J]. Applied and Environmental Microbiology, 2005, 71(12):8818-8824.
[47]Toymentseva AA, Schrecke K, Sharipova MR, et al. The LIKE system, a novel protein expression toolbox for Bacillus subtilis based on the liaI promoter [J]. Microbial Cell Factories, 2012, 11(1):143.
[48]Phan TTP, Schumann W. Development of a glycine-inducible exp-ression system for Bacillus subtilis [J]. Journal of Biotechnology,2007, 128(3):486-499.
[49]Heravi KM, Wenzel M, Altenbuchner J. Regulation of mtl operon promoter of Bacillus subtilis:requirements of its use in expression vectors [J]. Microbial Cell Factories, 2011, 10:83.
[50]Yang MM, Zhang WW, Chen YL, et al. Development of a Bacillus subtilis expression system using the improved Pglv promoter [J]. Microbial Cell Factories, 2010, 9(1):55.
[51]Panahi R, Vasheghani-Farahani E, Shojaosadati SA, et al. Induction of Bacillus subtilis expression system using environmental stresses and glucose starvation [J]. Annals of Microbiology, 2014, 64(2):879-882.
[52]Blom EJ, Ridder AN, Lulko AT, et al. Time-resolved transcriptomics and bioinformatic analyses reveal intrinsic stress responses during batch culture of Bacillus subtilis [J]. PLoS One, 2011, 6(11):e27160.
[53]Nijland R, Lindner C, Van Hartskamp M, et al. Heterologous production and secretion of Clostridium perfringens β-toxoid in closely related Gram-positive hosts [J]. Journal of Biotechnology,2007, 127(3):361-372.
[54]Jan J, Valle F, Bolivar F, et al. Construction of protein overproducer strains in Bacillus subtilis by an integrative approach [J]. Applied Microbiology and Biotechnology, 2001, 55(1):69-75.
[55]Lee SJ, Pan JG, Park SH, et al. Development of a stationary phasespecific autoinducible expression system in Bacillus subtilis [J]. Journal of Biotechnology, 2010, 149(1):16-20.
[56]Wenzel M, Müller A, Siemann-Herzberg M, et al. Self-inducible Bacillus subtilis expression system for reliable and inexpensive protein production by high-cell-density fermentation [J]. Applied and Environmental Microbiology, 2011, 77(18):6419-6425.
[57]Gat O, Inbar I, Aloni-Grinstein R, et al. Use of a promoter trap system in Bacillus anthracis and Bacillus subtilis for the development of recombinant protective antigen-based vaccines [J]. Infection and Immunity, 2003, 71(2):801-813.
[58]Harwood CR, Williams DM, Lovett PS. Nucleotide sequence of a Bacillus pumilus gene specifying chloramphenicol acetyltransferase[J]. Gene, 1983, 24(2):163-169.
[59]Wang J, Ai X, Mei H, et al. High-throughput identification of promoters and screening of highly active promoter-5’-UTR DNA region with different characteristics from Bacillus thuringiensis [J]. PLoS One, 2013, 8(5):e62960.
(責任編輯 狄艷紅)
Research Progress of Bacillus subtilis Expression System and Its Promoter Regulatory Elements
Yu Xiaoxia Tian Jian Liu Xiaoqing Wu Ningfeng
(Institute of Biotechnology, Chinese Academy of Agricultural Sciences, Beijing 100081)
As a Gram-positive bacteria, Bacillus subtilis is an attractive host for the production of heterologous secretory proteins for several reasons:it is non-pathogenic and the capable of secreting functional extracellular proteins directly to the culture medium, a great deal of vital information concerning large scale fermentation and production technology. One of the key factors for achieving high-level expression of heterologous proteins is the use of a strong and control promoter. In present, the promoters of Bacillus subtilis can be classified into three categories:constitutive promoters, inducer-specific promoters and autoinducible promoters. This paper described the advantages and disadvantages of Bacillus subtilis expression system and the classification of promoters. At the same time, we summarized the methods of the amplification of new promoters, which provided a foundation for improving Bacillus subtilis expression systems and the industrial production of heterologous proteins .
Bacillus subtilis;expression system;constitutive promoters;inducible promoters
10.13560/j.cnki.biotech.bull.1985.2015.02.005
2014-07-10
國家“863計劃”項目(2014AA021303)
余小霞,碩士研究生,研究方向:微生物分子生物學與基因工程;E-mail:yuxiaoxia198921@sina.com
伍寧豐,研究員,研究方向:微生物分子生物學與基因工程;E-mail:wuningfeng@caas.cn
