浸漬方法對CuO-CeO2/Cord.整體式催化劑性能的影響*
2015-11-28 10:39:14灣麗娟張紅霞
合成材料老化與應用 2015年4期
關鍵詞:催化劑
灣麗娟,張紅霞,呂 莉
( 內蒙古工業大學化工學院,內蒙古呼和浩特010051)
隨著環境保護的發展和需要,燃料電池汽車以其零排放或接近于零排放的特質[1],已經成為當今世界能源與交通領域開發的熱點。質子交換膜燃料電池(PEMFC)是目前最具應用前景的燃料電池,其以氫氣為燃料。氫氣的主要來源之一是通過水煤氣變換反應制取[2]。實驗證明,水煤氣變換反應制取的重整氣中通常會含有1%的CO,微量的CO會使質子交換膜燃料電池的性能急劇下降,因此必須將1%的CO 脫除到100mg/kg 以下。富氫氣中CO 優先氧化反應是將重整氣中CO 由10mL/L降到10μL/L 以下最有效的方法[3-5]。作為選擇性氧化催化劑,非貴金屬催化劑CuO-CeO2表現出良好的CO 選擇性和催化氧化活性,并逐漸成為該領域的研究熱點[6]。在前期研究中,已經成功制備出對富氫中CO 具有較高活性和選擇性的顆粒狀CuO-CeO2催化劑。但是顆粒狀的催化劑在使用過程中存在很多弊端,如高的床層壓降、氣體溝流、床層堵塞等,因此有必要進一步研究整體式催化劑[7-9]。
堇青石具有接近于零的熱膨脹系數[10],因此本課題選取堇青石為載體,研究載體的浸漬方法對CuO-CeO2/Cord. 催化劑的活性和選擇性進行了研究。
1 實驗部分
1.1 載體預處理
將大塊蜂窩狀堇青石打磨成大小適當的長方體做為催化劑載體。先用蒸餾水充分洗滌堇青石顆粒表面,以除去研磨時殘留的粉末。用20%的草酸溶液沸煮處理2h 后,再用蒸餾水洗滌幾次。在100℃的條件下干燥2h,于300℃焙燒3h 以除去附著的草酸。預處理后的載體進行活性組分的負載。
1.2 催化劑的制備
1.2.1 分步浸漬法(Step impregnation)
稱取適量的Ce(NO3)3溶解在適量去離子水中,配制成Ce(NO3)3溶液。采用飽和浸漬法將預處理好的堇青石載體浸入Ce(NO3)3溶液中,在室溫下陳化4h ~6h 后,于100℃干燥10h ~12h,然后在500℃的條件下焙燒4h。稱取適量的Cu(NO3)2溶解在適量的去離子水中,配制成Cu(NO3)2溶液。將冷卻后負載過CeO2的載體浸入Cu(NO3)2溶液,在室溫下陳化4h ~6h,于100℃干燥10h ~12h,然后在500℃的條件下焙燒4h,所得到的催化劑記為CuO/CeO2/Cord. (SI)。
1.2.2 共浸漬法(Co-impregnation)
稱取適量的Cu(NO3)2和Ce(NO3)3溶解在適量去離子水中,配制成Cu(NO3)2和Ce(NO3)3的混合溶液。采用飽和浸漬法將預處理好的堇青石載體浸入混合溶液中,在室溫下陳化4h ~6h,于100℃干燥10h ~12h,然后在500℃的條件下焙燒4h,所得到的催化劑記為CuO/CeO2/Cord. (CI)。
2 結果與討論
2.1 催化性能的測試
催化劑的活性和選擇性測試是在常壓下于固定床反應器進行。反應器是由石英制成的U 型管,測試時將U 型管裝在加熱爐中,爐溫是由放置于反應器外表面的熱電偶并通過LU -900 溫控儀來控制,測試氣體向下通過裝有催化劑床層的反應器。反應氣組成為(體積分數):50% H2+ 1% O2+1%CO+N2平衡。空速為40000mL·g-1·h-1。采用島津GC-8A 氣相色譜儀進行反應器出口在線分析,TCD 檢測。
圖1、圖2 分別為采用分步浸漬和共浸漬法制備的催化劑的活性-溫度和選擇性-溫度曲線。


圖1 分步浸漬和共浸漬制得的催化劑活性對比Fig.1 Activity contrast of catalysts made by separate impregnation and co-impregnation

圖2 分步浸漬和共浸漬制得的催化劑選擇活性對比Fig.2 Selectivity contrast of catalysts made by separate impregnation and co-impregnation
從圖1 中可以看出,采用共浸漬法制得的催化劑的活性高于采用分步浸漬法制得的催化劑的活性,共浸漬法制得的催化劑的T50在90℃左右,T100在115℃;而分步浸漬法制得的催化劑的T50在125℃左右,T100在155℃。這說明采用共浸漬法制得的催化劑中CuO 的分散效果要好于采用分步浸漬法制得的催化劑中CuO 的分散效果。由于采用共浸漬制備催化劑時,將Cu(NO3)2、Ce(NO3)3同時溶解于溶液中,有利于銅離子進入CeO2晶格,提高了CuO 的分散性,增加了活性位的可能,從而提高了催化劑的催化活性。
從圖2 中可以看出,采用分步浸漬法制得的催化劑的選擇性要優于采用共浸漬法制得的催化劑。采用分步浸漬法制得的催化劑的選擇性在95℃時開始下降,但下降速度緩慢,而采用共浸漬法制得的催化劑的選擇性從115℃時開始急劇下降。這可能是由于共浸漬法使得CuO 更好的分布在CeO2上從而使催化劑具有更高的催化活性,在促進CO 氧化的同時也促進了H2的氧化。當反應溫度超過115℃后,H2的消耗量開始增大,使得催化劑的催化選擇性下降,但采用共浸漬法制得的催化劑選擇性在150℃仍能達到90%。
2.2 XRD 研究
德國BRUBER 公司生產的D8 advance 型X -射線衍射儀。以Cu Kα光源(λ =0.154nm),管電壓為40kV,管電流為100mA,連續掃描速度為6°/min,掃描步長為0.04°,掃描范圍2θ=10° ~80°。
為了研究CuO 在催化劑中X 射線特征峰的表現狀況,我們先選取前期工作者用沉積沉淀法制備的有較好催化性能的CuO-CeO2催化劑和CuO、CeO2進行XRD 譜圖對比。三種物質的XRD 圖譜見圖3所示。在CuO 的XRD 譜中可以看到CuO 有眾多特征吸收峰,兩個最強峰分別位于2θ =35.5°、38.5°,而這些強峰在CuO-CeO2的XRD 譜中根本看不到,看到的僅是CuO-CeO2和CeO2具有近乎完全相同的XRD 譜。這說明當CuO 高度分散于CeO2的表面時,在催化劑的XRD 圖譜上將觀察不到CuO 的特征衍射峰。

圖3 CuO、CeO2、CuO-CeO2 XRD 圖譜對比Fig.3 XRD patterns contrast of CuO,CeO2 and CuO-CeO2
圖4 為CuO-CeO2、Cordierite 的XRD 譜圖與分步浸漬的CuO-CeO2/Cord. 和共浸漬的CuO-CeO2/Cord. 的XRD 譜圖對比。
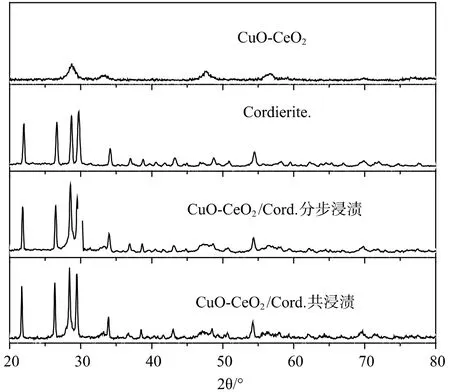
圖4 CuO-CeO2、Cordierite、CuO-CeO2/Cord. 分步浸漬與共浸漬XRD 譜圖對比Fig.4 XRD patterns contrast of CuO-CeO2,Cordierite and CuO-CeO2/Cord. made by separate impregnation and co-impregnation
從圖4 中可以看出,如沉積沉淀法制備的CuOCeO2催化劑XRD 譜圖所示,在圖譜中觀察不到CuO的特征衍射峰,觀察到CeO2的特征衍射峰分別位于2θ=28.5°、33.1°、47.5°、56.5°。在兩種不同方法制備的CuO-CeO2/Cord. 催化劑與堇青石的XRD 圖譜比較中我們發現,兩種催化劑的XRD 圖譜均在2θ=33.1°、47.5°、56.5°位置較堇青石的有明顯的增強,而這正是CeO2的特征衍射峰。除此之外沒有發現CuO 的特征峰,說明兩種制備方法都使得CuO高度分散于催化劑的表面。
3 結論
制備方法對CuO-CeO2/Cord. 整體催化劑中活性組分的分布有一定影響,活性組分的高度分散是整體催化劑具有優良催化性能的保證。結果表明,采用共浸漬法制得的催化劑活性明顯好于分步浸漬法,而且采用共浸漬法制得的催化劑在150℃時仍能保持90%的選擇性。
[1]衣寶廉. 燃料電池——原理·技術·應用[M].第一版. 北京:化學工業出版社,2003:160-161.
[2]A. K. Shukla,M. K. Ravikumar,K. S. Gandhi.Direct methanol fuel cells for vehicular applications[J]. Solid state electrochemistry,1998,2:117 -122.
[3]Olga Korotkikh,Robert Farrauto. Selective catalytic oxidation of CO in H2:fuel cell applications[J]. Catalysis Today,2000,62:249 -254.
[4]Jeong Kwi Seong,Oh Byeong Soo. Fuel economy and life-cycle cost analysis of a fuel cell hybrid vehicle[J]. Journal of Power Sources,2002,105(1):58 -65.
[5]Gregor Sedmak,Stanko Hocevar,and Janez Levec.Kinetics of selective CO oxidation in excess of H2over the nanostructured Cu0.1Ce0.9O2-ycatalyst[J]. Journal of Catalysis,2003,213:135 -150.
[6]Rui Lin,Meng-Fei Luo,Yi-Jun Zhong,et al. Comparative study of CuO/CeO.7Sn0.3O2,CuO/CeO2and CuO/SnO2catalysts for low-temperature CO oxidation [J]. Applied Catalysis A:General,2003,255:331 -336.
[7]M J Kahlich. Kinetics of the selective CO oxidation in H2-rich gas on Pt/Al2O3[J]. Journal of Catalysis,1997,171:93 -105.
[8]Friedrich M Hoffmann,Daniel J Dwyer[J]. Surface Science of Catalysis,1991:10 -17.
[9]Hisanori Tanaka,Shin-ichi Ito,Satoshi Kameoka,et al. Catalytic performance of K-promoted Rh/USY catalysts in preferential oxidation of CO in rich hydrogen[J]. Applied Catalysis A:General,2003,250:255 -263.
[10]朱洪法. 催化劑載體[M]. 北京:化學工業出版社,1980:27 -31.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
