快速高效miRNA分析流程在黃顙魚中的應用
2016-02-07 13:21:17王平平盧建國王樂欒培賢王秋實張曉峰
水產學雜志 2016年5期
關鍵詞:分析
王平平,盧建國,王樂,欒培賢,王秋實,張曉峰
(中國水產科學研究院黑龍江水產研究所,黑龍江 哈爾濱 150070)
快速高效miRNA分析流程在黃顙魚中的應用
王平平,盧建國,王樂,欒培賢,王秋實,張曉峰
(中國水產科學研究院黑龍江水產研究所,黑龍江 哈爾濱 150070)
高通量測序技術速度快、成本低、通量高,廣泛應用于miRNA領域研究。本研究基于高通量測序技術所產生的海量小RNA數據,結合已有的數據分析軟件,開發了一套快速高效一鍵化的miRNA分析流程。該流程整合多個生物信息學數據分析軟件,對多個miRNA高通量測序數據集進行標記、整合和去冗余分析,只需運行一次核心程序就可以實現對多個miRNA高通量測序數據的分析,避免每個樣本單獨數據分析的技術重復,精簡后的數據集能大幅度減少軟件計算量,顯著提高軟件運行效率。本研究利用快速高效miRNA分析流程分析黃顙魚性腺XX卵巢、XY精巢、YY精巢的miRNA高通量測序數據,獲得一批準確的黃顙魚保守miRNA。在相同參數設置下,miRNA分析流程可以顯著節約分析時間。該流程最終輸出結果為多樣本整合后的miRNA表達數據,便于研究者直接進行樣本之間的比較和miRNA的表達差異,減少研究者手動整合分析結果的操作步驟。miRNA分析流程針對多樣本miRNA測序數據具有明顯的優勢,樣本越多測序量越大,軟件運行效率越高。針對日益積累的海量小RNA測序數據,miRNA分析流程高效快速一鍵化數據處理優勢將會越來越明顯。
miRNA;高通量測序;miRNA識別方法;黃顙魚
microRNAs(miRNAs)是一類內源性非編碼RNA,長度約為18~30個核苷酸。miRNA基因首先在RNA聚合酶的作用下轉錄成初始轉錄本,經Drosha酶剪切形成miRNA前體,再進一步由Dicer酶切割產生成熟體miRNA[1]。成熟miRNA通過堿基互補配對的方式識別靶基因的mRNA,降解靶基因mRNA或抑制靶基因的翻譯。miRNA參與生物體內多種調控通路,包括發育、器官形成、細胞增殖和凋亡等[2]。因此準確識別miRNA,了解其生物功能具有重要科學意義。
高通量測序技術測序速度快、成本低、通量高,已廣泛應用于miRNA領域研究。miRNA高通量測序可以從系統的全局的角度獲得生物體內絕大部分表達的miRNA,廣泛用于比較不同發育階段、不同組織及不同條件下的miRNA表達譜。隨著miRNA高通量測序數據的不斷豐富,使得快速高效的miRNA識別方法越來越受關注。
miRDeep是一款被廣泛使用的miRNA深度測序數據分析軟件,由Friedlander團隊在2008年發表[3],2012年更名為miRDeep2[4]。miRDeep主要基于貝葉斯概率模型來預測miRNA,數據分析過程[5]包括BLAST[6,7]或Bowtie[8]比對測序數據到參考基因組、篩選得到候選miRNA前體序列、前體序列打分等,最終獲得物種保守的miRNA。miRDeep在無基因組注釋信息的情況下,在真渦蟲樣本上仍能達到86%的敏感度[3]。miRDeep2在時間效率和內存分配上都有很大的改進,能同時識別正義和反義鏈上的miRNA,且允許一個或多個位置的堿基錯配。miRDeep2預測動物miRNA的準確率可以達到98.6%~99.9%[4]。miRDeep2已經被廣泛用于miRNA高通量測序數據的分析[9-12]。
miRDeep2軟件操作簡單,缺點是多樣本數據需要每個樣本單獨進行分析,分析結果還需要繁瑣的整合過程。目前的研究很少只對單一樣本進行小RNA測序,通常都需要對多個樣本小RNA測序數據進行對比性研究。針對于多樣本miRNA測序數據,miRDeep2結果的整合過程需要一定的編程基礎和生物信息學知識,在這一整合過程中需耗費較多的時間和精力。本研究基于高通量測序技術所產生的海量小RNA數據,開發出一套基于miRDeep2的多樣本整合分析流程,只需運行一次核心程序就可以實現對多個miRNA高通量測序數據的處理和分析,極大地節約數據分析的時間和精力。
1 材料與方法
1.1 材料
本研究開發的miRNA快速識別流程適用于Illumina高通量測序平臺獲得的小RNA測序數據,輸入數據為測序reads的fasta格式文件。Fastq格式的原始測序數據可以根據本流程中提供的軟件和本實驗室開發的腳本,按自身需求進行個性化的數據預處理,包括測序數據接頭序列去除、數據質量控制和格式轉換。
本研究使用的測試數據為Illumina測序平臺的黃顙魚Pelteobagrus fulvidraco miRNA高通量測序數據,包含雌魚XX卵巢、雄魚XY精巢和全雄魚YY精巢三個樣本的小RNA高通量測序數據[13]。數據來自NCBI的SRA數據庫,訪問編號分別為SRR1154617、SRR1154615和SRR1154616。
1.2 miRNA高通量測序數據分析流程
miRNA快速高效識別流程是以開放軟件miRDeep2為核心,通過整合多個數據分析軟件和實驗室自主開發的腳本程序,達到快速高效一鍵化的分析miRNA高通量測序數據的目的。該流程命名為miRDeep-pipeline,主要步驟(圖1)包括:原始高通量測序數據預處理;多樣本數據標記、整合和聚類分析;miRDeep2軟件分析;多樣本數據分析結果提取和比較。
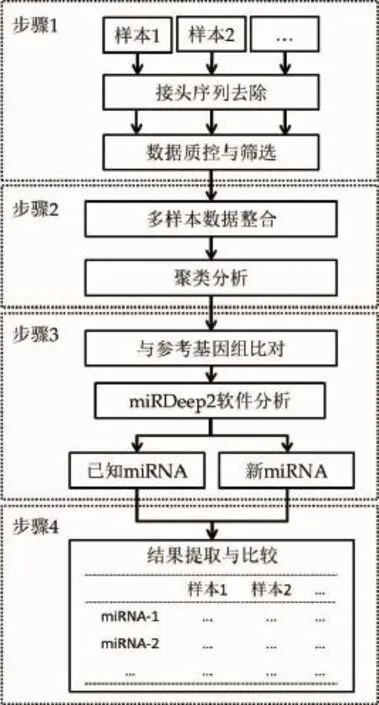
圖1 miRDeep-pipeline流程圖Fig.1 The flowing chart of miRDeep-pipeline
miRDeep-pipeline的輸入數據為測序reads的fasta格式數據,其他格式的高通量測序數據,如sra、fastq等格式數據。本流程中也提供相應的分析軟件和實驗室自主開發的腳本程序,研究者可以根據自身需求進行個性化的原始數據預處理。miRD-eep-pipeline整合的數據分析軟件和實驗室自主開發的腳本程序見表1,包括SRAToolkit、Cutadapt[14]等。實驗室自主開發的腳本程序miRNA_length_ stats.pl、stats_nuc_bias.pl,主要用于統計分析miRNA測序數據,包括miRNA長度分布統計和堿基偏好性統計。miRDeep-pipeline提供Illumina測序平臺小RNA測序數據通用的接頭序列,適用于大多數Illumina平臺,研究者也可以根據自身數據特點選擇合適的接頭序列處理數據。mirdeep-pipeline是本流程的核心程序,可以實現一鍵化的多樣本miRNA高通量測序數據分析。
1.3 miRDeep-pipeline核心數據處理策略
miRDeep-pipeline的核心內容是標記、整合和聚類分析多樣本數據,減少數據分析過程中重復的軟件調用,精簡的數據集也能有效減少軟件運行時間。根據不同的樣本標簽以及序列重復次數標簽可以有效地拆分miRDeep2輸出結果,達到快速高效分析多樣本數據的目的。
針對多個樣本單獨分析過程中重復調用miRDeep2軟件造成的時間和精力浪費,miRD-eep-pipeline首先標記多個樣本數據,整合標記后的數據集;然后根據序列相似性對測序數據進行聚類分析和序列重復次數標記,減少重復序列重復運算的計算資源和計算時間的浪費,獲得精簡的待分析數據集。在保證結果準確性的前提下,所有樣本中重復的序列只需要進行一次基因組比對和miRD-eep2軟件計算,大大提高了軟件運行效率;最后根據不同的樣本標簽以及序列重復次數標簽拆分miRDeep2輸出結果,樣本標簽用于區分來自不同樣本的分析結果,序列重復次數標簽用于估計每一個識別的miRNA在各樣本中的表達量。最終獲得每個樣本中表達的miRNA,及其對應的表達量。
miRDeep-pipeline一方面大大減少多個樣本在單獨數據處理過程中頻繁的軟件調用;另一方面,結合多樣本數據標記、整合和聚類分析策略,大大減少軟件計算的數據量,節約計算成本。另外,miRDeep-pipeline的輸出結果可以直接比較不同樣本,分析差異表達的miRNA,避免對每個樣本單獨進行miRDeep2計算后繁瑣的數據合并工作,提高了工作效率。miRDeep-pipeline整個運行過程中只需要調用一次核心程序“mirdeep-pipeline”就可以實現對全部數據的處理和結果整合分析,真正實現一鍵化的操作流程。

表1 miRDeep-pipeline整合的軟件及實驗室自主開發的腳本Tab.1 The list of software and laboratory developed scripts integrated in miRDeep-pipeline
2 結果與分析
2.1 軟件效率
用miRDeep2分別處理黃顙魚XX卵巢、XY精巢、YY精巢三個樣本的小RNA高通量測序數據,并與miRDeep-pipeline進行對比(表2)。miRD-eep-pipeline一鍵化地處理黃顙魚三個樣本小RNA測序數據,大概需要7h。用miRDeep2分別處理XX、XY和YY三個樣本程序運行總時間大概為13.5h,單獨處理樣本XY需要將近6h,如果可以并行化處理,那么miRDeep2數據分析總時間需要將近6h。在本文中,miRDeep-pipeline相對于單線程運行miRDeep2可以節約一半的軟件運行時間,與多線程運行miRDeep2相比無明顯優勢。但是miRDeep-pipeline的主要優勢在于其可以實現一鍵化分析多個樣本數據及整合結果,大大減少軟件的重復調用和多樣本分析結果繁瑣的整合步驟,節約了時間,提高了效率。
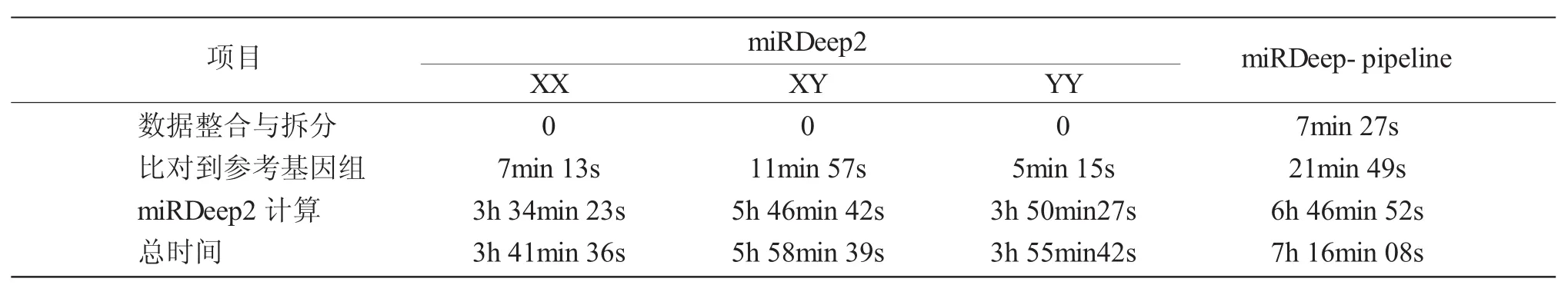
表2 miRDeep-pipeline運行時間效率Tab.2 Time efficiency of miRDeep-pipeline process
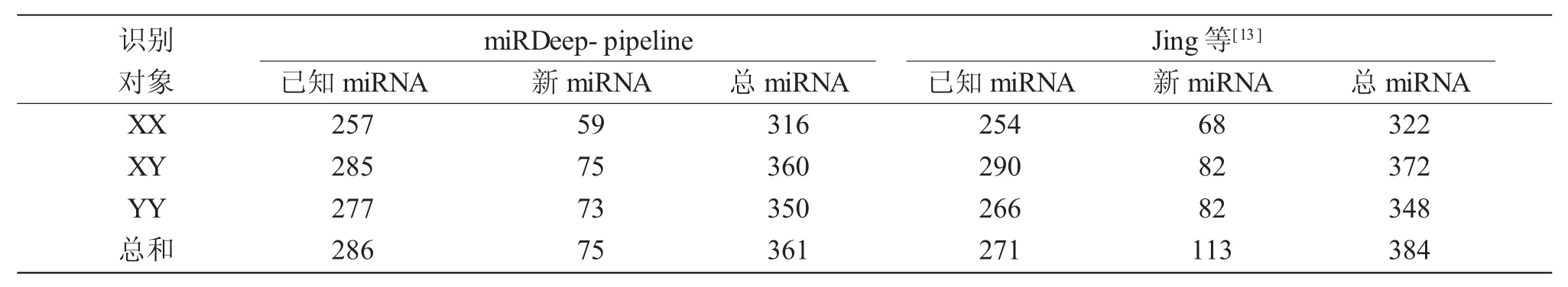
表3 黃顙魚miRNA的識別與比較Tab.3 Identification and comparison of miRNA in yellow catfish
2.2 黃顙魚miRNA的識別與比較
miRDeep-pipeline在黃顙魚XX卵巢,XY精巢和YY精巢樣本中共識別543個保守miRNA,其中能在黃顙魚基因組上準確定位并且pre-miRNA能形成完整的莖環結構的有361個(表3)。在高通量測序數據和黃顙魚基因組的雙重支持下,獲得的361個黃顙魚保守的miRNA具有較高的可信度,其中286個為miRBase數據庫[15,16]中收錄的已知miRNA,剩余75個為新miRNA。在XX卵巢、XY精巢和YY精巢中分別識別出316、360和350個保守miRNA,其中新miRNA數目分別為59個、75個和73個。Jing等人[13]在2014年利用商業軟件ACGT101-miR v4.2分析這部分數據的結果與 miRD-eep-pipeline的預測結果具有較好的一致性。
Jing等通過實時定量PCR技術驗證部分miRNA,這些miRNA在本研究中也被正確識別和定量(圖2)。從圖2可以看出,miRDeep-pipeline對表達量的估計與商業軟件ACGT101-miR v4.2具有較好的一致性。
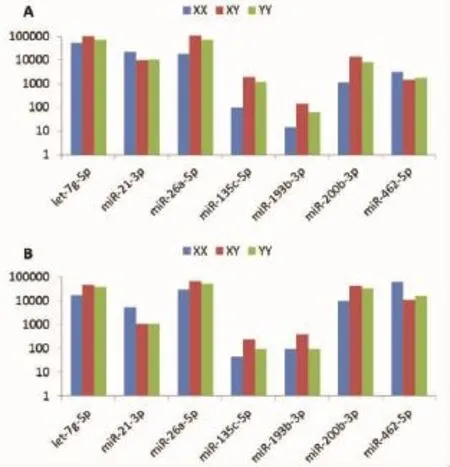
圖2 部分miRNA定量結果Fig.2 The expression level of several selected miRNAs
3 討論
隨著測序技術的發展,獲取一組miRNA高通量測序數據越來越容易,高效快速的一鍵化的處理軟件越來越受歡迎。本研究整合了一整套miRNA高通量測序分析軟件,合理設計算法策略,巧妙地先合并后拆分多個樣本數據,達到快速、高效、一鍵化的處理多樣本miRNA高通量數據的目的。
miRDeep-pipeline整合了優秀的miRDeep2軟件[4],具有與miRDeep2同等的精確性和優于miRDeep2的運行效率。
首先在操作上,單純miRDeep2針對多個樣本需要重復相同的分析操作,而miRDeep-pipeline可以一次性處理多個樣本,并且只需要運行一個核心程序mirdeep-pipeline,就可以完成對多個miRNA測序數據的分析工作,大大精簡了操作步驟。
其次在軟件運行成本上,miRDeep-pipeline對多個樣本數據進行先合并后拆分的策略,在合并過程中進行聚類分析,避免重復序列的重復計算。在黃顙魚性腺miRNA測序數據上試驗,miRD-eep-pipeline相對于單線程運行miRDeep2軟件至少可以節約一半的分析時間。miRDeep-pipeline處理多個樣本的時間優勢會隨著數據量的增加而更加明顯,因為生物體內存在的miRNA是有限的,測序深度的增加和樣本數的增加對數據復雜度的增加是很有限的,大多數情況下只會影響序列的重復次數,對唯一序列的總數影響非常微弱[17]。在本研究中,對序列進行合并后去冗余,精簡后的數據集在測序深度和樣本數增加的情況下增加幅度相對微弱,不會導致大幅度的軟件計算時間的增加,因而miRDeep-pipeline在處理大數據和多樣本時隨著數據量的增加而效率更高,優勢更明顯。
最后,miRDeep-pipeline最終輸出結果為多樣本整合后的miRNA表達數據,便于直接進行樣本之間比較和miRNA差異表達分析,減少手動整合多樣本分析結果的操作,節約大量時間成本,提高工作效率。沒有任何編程基礎的研究者也很容易讀懂分析結果,開展進一步的研究工作。
本研究開發的miRNA快速高效分析流程針對多樣本miRNA高通量測序數據具有明顯的優勢,樣本數量越多測序量越大,軟件運行效率越高。針對日益積累的海量小 RNA測序數據,miRD-eep-pipeline高效快速一鍵化數據處理流程將被廣泛使用。
[1]Chen X.Small RNAs and their roles in plant development[J].Annual Review of Cell and Developmental Biology, 2009,25(1):21-44.
[2]Bartel D P.MicroRNAs:genomics,biogenesis,mechanism, and function[J].Cell,2004,116(2):281-297.
[3 Friedl?nder M R,Chen W,Adamidi C,et al.Discovering microRNAs fromdeep sequencingdata usingmiRDeep[J]. Nat Biotechnol,2008,26(4):407-415.
[4]Friedl?nder M R,Mackowiak S D,Li N,et al.miRDeep2 accurately identifies known and hundredsofnovel microRNA genes in seven animal clades[J].Nucleic Acids Res,2012,40(1):37-52.
[5]萬琳霞,丁建棟,關佶紅.計算方法預測microRNA研究進展[J].計算機應用與軟件,2012,29(5):159-162,194.
[6]Altschul S F,Gish W,Miller W,et al.Basic local alignment search tool[J].J Mol Biol,1990,215(3):403-410.
[7 Mount D W.Using the basic local alignment search tool(BLAST)[J].CSH Protocols,2007(14):pdb.top17,doi: 10.1101/pdb.top17.
[8 Langmead B,Trapnell C,Pop M,et al.Ultrafast and memory-efficient alignment ofshort DNA sequences to the human genome[J].Genome Biology,2009,10(3):R25.
[9]Xu F,WangX,FengY,et al.Identification ofconserved and novel microRNAs in the Pacific oyster Crassostrea gigas by deep sequencing[J].PLoSOne,2014,9(8):e104371.
[10 Li R,Beaudoin F,Ammah A A,et al.Deep sequencing shows microRNA involvement in bovine mammary gland adaptation to diets supplemented with linseed oil or safflower oil[J].BMCGenomics,2015,16(1):884.
[11]Fan G,CaoX,Niu S,et al.Transcriptome,microRNA,and degradome analyses of the gene expression of Paulownia with phytoplamsa[J].BMCGenomics,2015,16(1):896.
[12 Keller A,Leidinger P,Meese E,et al.Next-generation sequencing identifies altered whole blood microRNAs in neuromyelitis optica spectrum disorder which may permit discriminationfrommultiplesclerosis[J].Journalofneuroinflammation,2015,12(1):196.
[13]Jing J,Wu J,Liu W,et al.Sex-biased miRNAs in gonad and their potential roles for testis development in yellow catfish[J].PloSONE,2014,9(9):e107946.
[14 Martin M.Cutadapt removes adapter sequences from high-throughput sequencing reads[J].EMBnet Journal, 2011,17(1):10-12.
[15]Griffiths-JonesS,GrocockRJ,vanDongenS,etal.miRBase: microRNA sequences,targets and gene nomenclature[J]. NucleicAcidsRes,2006,34:D140-D144.
[16 Kozomara A and Griffiths-Jones S.miRBase:annotating high confidence microRNAs using deep sequencing data[J].Nucleic Acids Res,2014,42:D68-D73.
[17]Sims D,Sudbery I,Ilott N E,et al.Sequencing depth and coverage:key considerations in genomic analyses[J].Nature Reviews Genetics,2014,15(2):121-132.
Rapid and Efficient miRNA Identification Pipeline and its Application in Yellow Catfish
WANG Ping-ping,LU Jian-guo,WANG Le,LUAN Pei-xian,WANG Qiu-shi,ZHANG Xiao-feng
(Heilongjiang River Fisheries Research Institute,Chinese Academy of Fishery Sciences,Harbin 150070,China)
High-throughput sequencing technologies have been widely used in miRNA studies with the advantages of high speed,low cost and high throughput.Based on the massive miRNA data from high-throughput sequencing,we developed a rapid and efficient miRNA identification pipeline integrated with several data analysis softwares.By marking,merging and clustering of several miRNA sequencing data sets,our pipeline can avoid duplicate analysis processes for every single data set,which can greatly reduce the amount of calculation and significantly improve the software efficiency.Just run one more time of our pipeline,all the samples will be analyzed.In this study,we analyzed miRNA sequencing data from XX ovaries,XY testis and YY testis of yellow catfish(Pelteobagrus fulvidraco)with this pipeline,and identified a number of conserved miRNAs,with significant reduce in time in the same parameters by pipeline.The integrated results from our pipeline are comparable between samples and easily to do further miRNA differentially expression analysis,which will greatly reduce the manual integration operation for every single results.Our miRNA identification pipeline has obvious advantages in processing multiple data sets.The more samples and greater data sets,the higher the efficiency.The advantages of our pipeline will be more and more obvious with the increasing accumulated miRNA sequencing data.
miRNA;high-throughput sequencing;miRNA identification;yellow catfish(Pelteobagrus fulvidraco)
S917
A
1005-3832(2016)05-0027-05
2016-06-10
中央級公益性科研院所基本科研業務費專項資金(HSY201505).
王平平(1988-),女,碩士,研究實習員,從事魚類生物信息學和遺傳育種研究.E-mail:wangpingping@hrfri.ac.cn
猜你喜歡
現代畜牧科技(2021年9期)2021-10-13 06:39:14
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
當代經濟研究(2016年5期)2016-12-01 03:12:05
現代農業(2016年5期)2016-02-28 18:42:46
出版與印刷(2016年3期)2016-02-02 01:20:11
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
華北水利水電大學學報(社會科學版)(2014年3期)2014-04-16 04:38:31
終身教育研究(2014年5期)2014-02-28 01:23:06
