氣相色譜-串聯質譜法檢測大米中20種農藥殘留
2016-05-13 09:31:56董振霖孫玉玉尚寶玉劉佳成曲世超紀明山
分析測試學報 2016年4期
陳 溪,董振霖,孫玉玉,尚寶玉,劉佳成,曲世超,紀明山
(1.沈陽農業大學 植物保護學院,遼寧 沈陽 110866;2.大連出入境檢驗檢疫局 檢驗檢疫技術中心,
遼寧 莊河 116400;3.遼寧出入境檢驗檢疫局 檢驗檢疫技術中心,遼寧 大連 116001)
氣相色譜-串聯質譜法檢測大米中20種農藥殘留
陳溪1,2,董振霖3,孫玉玉3,尚寶玉2,劉佳成2,曲世超2,紀明山1*
(1.沈陽農業大學植物保護學院,遼寧沈陽110866;2.大連出入境檢驗檢疫局檢驗檢疫技術中心,
遼寧莊河116400;3.遼寧出入境檢驗檢疫局檢驗檢疫技術中心,遼寧大連116001)
摘要:建立了兩種可簡便、靈敏、快速、批量測定大米中20種農藥殘留的分析方法。樣品經乙腈提取后,使用C18固相萃取柱(SPE)和QuEChERS兩種不同凈化方法,采用氣相色譜-串聯質譜選擇反應監測(SRM)模式對20種農藥進行檢測,通過保留時間、選擇離子及其相對豐度定性,外標法定量。結果表明,20種農藥的線性范圍為0.01 ~0.2 μg/mL,相關系數均大于0.99。在0.01,0.05,0.20 mg/kg加標水平下,采用C18柱對大米樣品進行提取凈化,20種農藥的平均回收率為63.3%~96.0%,相對標準偏差(RSD)為1.9%~13.9%;采用QuEChERS方法對大米樣品進行提取凈化,20種農藥的平均回收率為67.7%~121.0%,RSD為1.9%~16.4%。此兩種方法無明顯差異,為農藥多殘留檢測提供了不同的思路,能夠對大批量大米樣品進行同時檢測,數據準確可靠,回收率與精密度均符合農藥殘留的分析要求。
關鍵詞:大米;農藥殘留;氣相色譜-串聯質譜(GC-MS/MS);QuEChERS;固相萃取(SPE)
大米是人們的主要糧食。1990年以來,得益于農藥及化肥的使用,水稻的生產率以每年0.4%的速度增長。雖然使用農藥能夠有效控制雜草與病蟲害,但農作物可能由此受到嚴重污染,因此對大米產品的農藥殘留監控關系著人們的身體安全與食品安全[1]。國際食品法典委員會(CAC)[2]、歐盟委員會(European Commission)[3]、美國環境保護署(USEPA)[4]等各國相關組織及部門紛紛建立了食品中最大殘留限量要求(MRLs),我國也于2014年實施了國家標準GB 2763-2014《食品中農藥最大殘留限量》,涉及農藥387種、檢測項目達3 650項[5]。
隨著農藥殘留標準的日益嚴格及農藥殘留檢測項目的不斷增多,建立簡便、快速、重現性高的農藥多殘留檢測方法迫在眉睫。目前常用的樣品前處理方法包括QuEChERS法[6-11]、凝膠滲透凈化色譜(GPC)[12-14]、固相萃取(SPE)[15-16]凈化方法;而農藥多殘留的檢測技術,也逐漸以質譜檢測技術為主導[17]。雖然氣相色譜(GC)、氣相色譜質譜聯用(GC-MS)技術在農藥殘留的檢測中有著重要作用,但GC法由于基質的干擾,常遇到某些農藥不易定性問題,可能會出現許多雜質峰或造成假陽性結果;GC-MS在一定程度上解決了GC法面臨的假陽性問題,但對復雜樣品的檢測仍存在嚴重的基質干擾情況,較難鑒定目標物的分子結構[18-20]。因此,高選擇性、高靈敏度、高分辨率的多農殘同時篩查定性和確證定量分析手段應運而生,其中氣相色譜-串聯質譜法(GC-MS/MS)較一級質譜有著更強的抗干擾能力,能夠實現農藥的定量與定性篩查分析[1,21-26]。
遼寧地區作為大米的主產地,大米產品主要出口日本、韓國等。目前,針對谷物的農殘檢測方法以GB/T 20770-2008《糧谷中486種農藥及相關化學品殘留量的測定 液相色譜-串聯質譜法》、GB/T 19649-2006《糧谷中475種農藥及相關化學品殘留量的測定 氣相色譜-質譜法》、SN/T 2149-2008《進出口食品中解草嗪、莎稗磷、二丙烯草胺等110種農藥殘留量的檢測方法 氣相色譜-質譜法》為主,而文中涉及的20種農藥,均為2014年新增的輸韓大米檢測項目,與GB/T 20770-2008相比有11種農藥在該標準中未涉及;與GB/T 19649-2006相比,有5種農藥在該標準中未涉及;與SN/T 2149-2008相比有12種農藥在該標準中未涉及。且迄今為止尚無氣相色譜-串聯質譜的檢測標準,該方法的建立對科研人員及一線檢測人員有一定借鑒意義,能夠為出口大米的檢驗和產品的質量控制提供科學依據和技術支持。
1實驗部分
1.1儀器及試劑
氣相色譜-串聯質譜儀(GC-MS/MS,美國Thermo公司),配電子轟擊源(EI);電子天平(AE 260,瑞士梅特勒公司);均質機(T 25 digital ULTRA-TURRAX,德國IKA公司);離心機(Z-323 k,德國赫默公司);旋轉蒸發器(R215-PROV-V700,BIBBY RE 100);UniElut C18EC(2 000 mg/12 mL,中國華譜Acchrom公司)。
標準品:克草敵、氟丙嘧草酯、氯甲酰草胺、溴丁酰草胺、二丙烯草胺、苯胺靈、九氯、六氯苯,購自德國Dr.Ehrenstorfer GmbH。環庚草醚、酞菌酯、氯酞酸甲酯、滅藻醌、毒草胺、乳氟禾草靈、炔苯酰草胺、二甲草胺、四溴菊酯、莠滅凈、撲滅津、戊環唑,購自美國Sigma-Aldrich Laborchemikalien GmbH。農藥標準品純度均大于98%。
乙腈、甲醇、丙酮(色譜純,美國Fisher公司);乙二胺-N-丙基硅烷(PSA)粉末(Bondesil-PSA,40 μm,美國瓦里安公司);C18粉末(Octadecyl,40 μm,美國J.T.Baker公司);無水硫酸鎂、NaCl(分析純,天津市科密歐化學試劑有限公司)。無水硫酸鎂在650 ℃下灼燒4 h,冷卻至120 ℃后,置干燥器中備用;實驗用水為高純水。
1.2標準溶液配制
標準儲備液:準確稱取20種農藥標準品各10 mg(精確至0.1 mg)分別置于10 mL容量瓶中,用丙酮溶解并定容至刻度,得到質量濃度為1 000 μg/mL的農藥標準儲備液,并儲存于4 ℃冰箱中。
混合標準儲備液:準確移取25 μL上述1 000 μg/mL標準儲備液于10 mL容量瓶中,用丙酮定容,得到質量濃度為2.5 μg/mL的混合標準儲備液,儲存于4 ℃冰箱中。
混合基質標準工作溶液:為避免基質效應,配制基質標準工作液作為定量依據。用陰性空白樣品提取液作為溶劑,得濃度分別為0.01,0.025,0.05,0.1,0.2 μg/mL的混合基質標準工作溶液。
1.3樣品前處理
1.3.1C18柱凈化法提取:稱取10.0 g(精確至0.1 g)磨碎的大米(過20目篩),置于100 mL離心管中,加入30 mL乙腈,勻漿3 min,4 000 r/min離心10 min,上清液轉移至雞心瓶,殘渣部分再加入20 mL乙腈,經勻漿離心后,合并提取液。真空濃縮至干(48 ℃,110 r/min),用5 mL乙腈溶解,待凈化操作。
凈化:將C18(UniElut C-18 EC,2 g/12 mL)SPE柱用10 mL乙腈預淋洗,取2.5 mL上述提取液上樣并收集,用20 mL乙腈分3次洗脫C18柱,合并后濃縮至干。用2 mL丙酮溶解,供GC-MS/MS檢測。
1.3.2QuEChERS法提取:稱取10.0 g(精確至0.1 g)磨碎的大米(過20目篩)至50 mL離心管中,加入10 mL高純水,浸泡30 min后加入10 mL乙腈、4 g無水MgSO4和1 g NaCl,振搖3 min,離心(4 000 r/min,5 min)。
凈化:在2 mL離心管中加入50 mg PSA、50 mg C18粉末和150 mg無水MgSO4,移取提取液1 mL,振蕩混勻1 min后離心(4 000 r/min,5 min),取上清液供GC-MS/MS檢測。
1.4GC-MS/MS條件
色譜柱:DB-5MS(30 m×0.25 mm i.d.×0.25 μm);載氣:氦氣,純度≥99.995%,流速:1.0 mL/min;色譜柱溫度:70 ℃保持1 min,以15 ℃/min升至230 ℃,再以30 ℃/min升至280 ℃,保持10 min;進樣口溫度:250 ℃;氣相色譜-串聯質譜接口溫度:250 ℃;進樣量:1 μL;進樣方式:無分流進樣,1.0 min后開閥;電離方式:EI;電離能量:70 eV;測定方式:選擇反應監測方式(SRM);溶劑延遲:3 min。每種化合物通過GC-MS/MS檢測的保留時間、離子對等相關參數見表1。
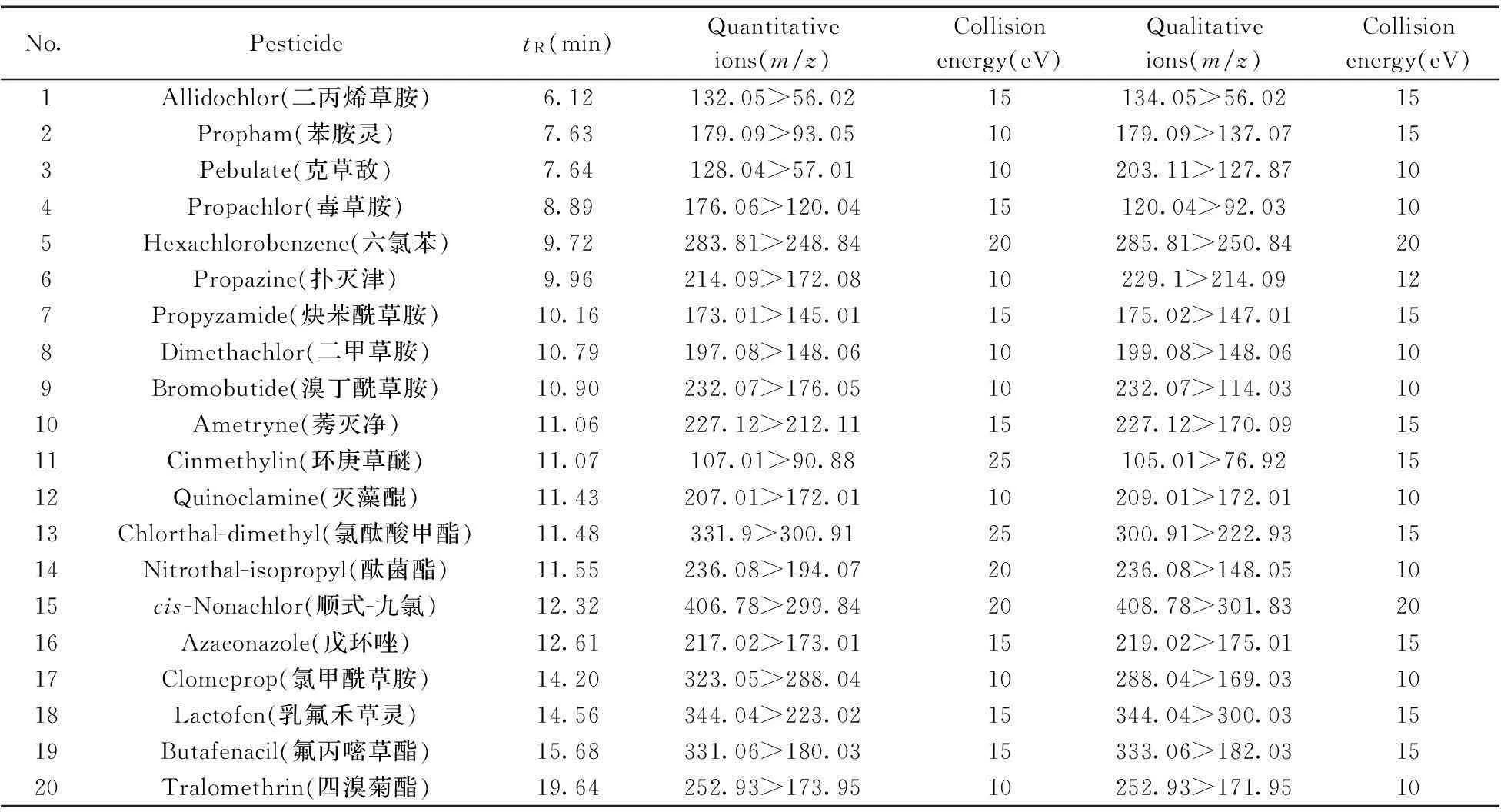
表1 20種農藥的質譜分析參數
2結果與討論
2.1提取溶劑的選擇
大米為干樣品,提取前需加水使其充分浸潤,以保證提取劑與樣品充分接觸。根據農藥的化學性質,選擇甲醇、丙酮、乙腈、二氯甲烷和乙酸乙酯作為提取溶劑,對大米樣品進行提取實驗,并對提取效果進行比較。結果表明,采用乙酸乙酯、二氯甲烷作為提取溶劑時,會提取出更多的脂肪類雜質,不利于后續凈化;甲醇作為提取劑時部分可溶性淀粉和水分會被一并萃取,導致旋轉蒸發的溫度過高,回收率偏低,且提取液被蒸干后,淀粉殘留在瓶壁上,增加了提取液轉移的難度,回收率結果不穩定。丙酮作為萃取劑蒸干后也會有水分殘留,需要增加除水的操作步驟,而且其對脂肪類油脂的萃取物也較多,同樣對回收率有較大影響。使用乙腈作提取劑時,對色素和基質中的蠟質、脂肪等非極性成分的提取能力相對較小。此外,乙腈能與水形成共沸物而將水分蒸出,無需除水,其提取效率更高。因此最終采用乙腈作為提取溶劑。
2.2凈化方法的研究
2.2.1C18柱洗脫液的選擇其他因素固定,采用不同比例的甲醇-乙腈溶劑作為洗脫液。結果顯示,隨著洗脫液中乙腈比例的上升,20種農藥的回收率不斷提高,故實驗選擇純乙腈作為洗脫溶劑。
2.2.2C18柱洗脫液體積的選擇其他因素固定,以不同體積的乙腈(5,10,20,30,40,50 mL)作為洗脫液,測定20種農藥的回收率。結果顯示,隨著洗脫液體積的增大,農藥的回收率呈先上升后下降的趨勢,其原因可能是由于洗脫液用量過多,旋蒸時間過長,造成了農藥的損失,反而使其回收率降低。當洗脫溶劑為20 mL時,20種農藥的萃取效果最好,其回收率均在70%~110%之間。
2.2.3QuEChERS法QuEChERS是一種快速、簡單、廉價、高效、耐用、安全的樣品前處理方法,參考AOAC 2007.01[27]、EN 15662[28]及文獻方法[29],對鹽析劑和吸水劑進行選擇。常用的鹽析劑為NaCl 和CH3COONa,吸水劑為無水Na2SO4和無水MgSO4。對于兩種鹽析劑而言,NaCl和CH3COONa對回收率無顯著影響,20種農藥的平均回收率相差在2.7%以內。而無水MgSO4的吸水能力更佳,在吸水過程中放熱,有利于農藥的提取;且無水MgSO4粒度更小,在振搖與渦旋的過程中可與樣品混合得更加充分,并通過與乙腈的協同作用,提高提取效率。因此,最終確定采用文獻方法[29],即加入1 g NaCl與4 g無水MgSO4。
另外,QuEChERS方法在凈化過程中,常采用PSA吸附劑除去樣品基質中的脂肪酸、有機酸和糖等雜質,石墨化炭黑(GCB)去除色素和甾醇,用C18反相吸附劑除去基質中的脂肪和脂類等非極性有機物,并通過加入無水MgSO4進一步除水,以提高農藥的萃取率。考察了添加50 mg PSA、50 mg C18或25 mg PSA、25 mg C18作為凈化劑時的凈化效果,結果表明,以50 mg PSA、50 mg C18為凈化劑時,基質效應相對較小,凈化效果較好,20種農藥的回收率為85.5%~106.4%。因此,本實驗在凈化過程中僅添加PSA及C18。
2.3質譜條件的優化
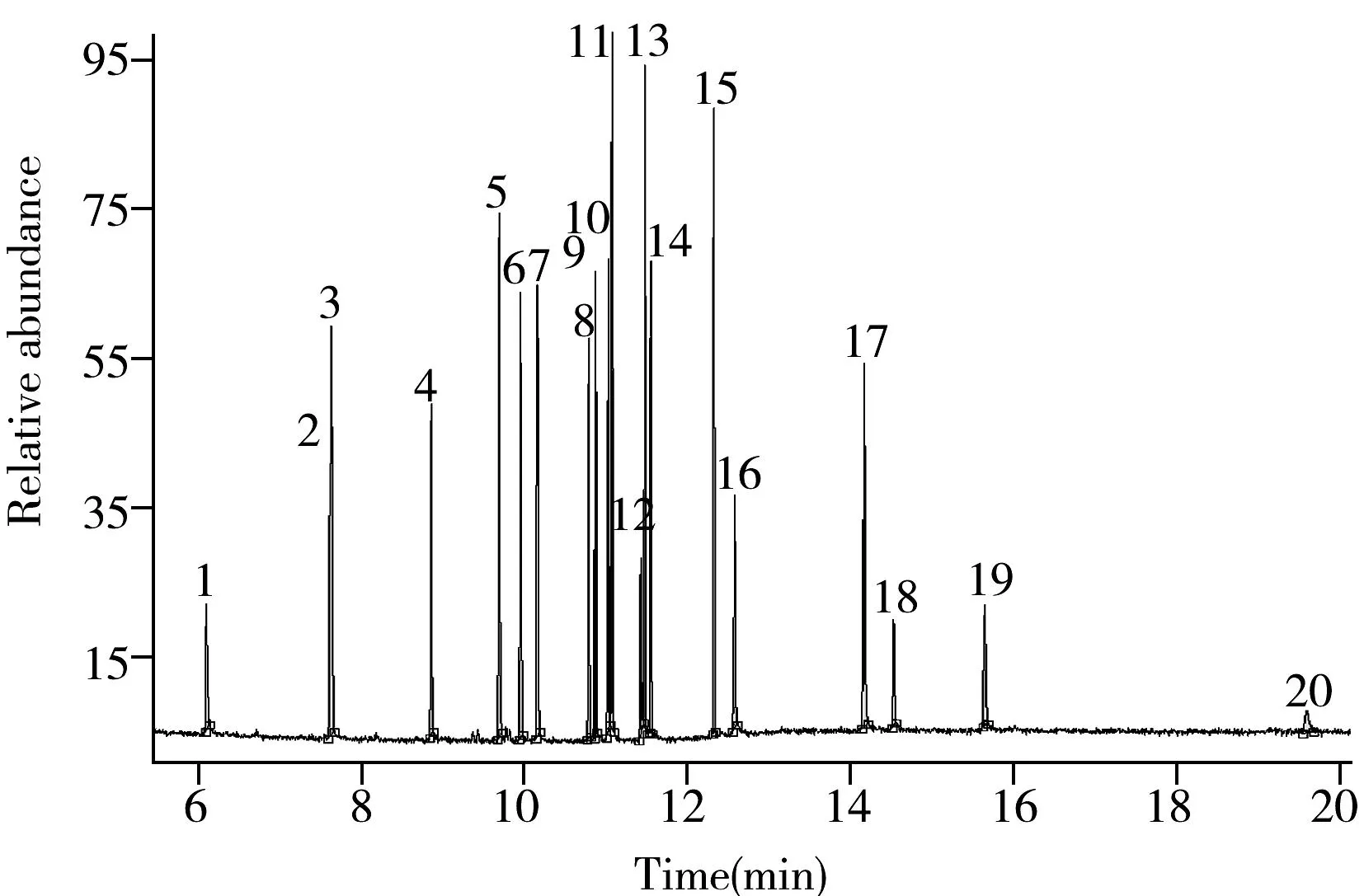
圖1 20種農藥的總離子流圖Fig.1 TIC of 20 pesticides standard solutionthe number represented is the same as that in Table 1
將1 μg/mL的20種農藥單標溶液進行單級質譜全掃描,選擇其中有代表性的母離子碎片;對每個母離子進行碰撞能優化,根據子離子掃描質譜圖,選取最佳的定性和定量離子對。最終確定各農藥的母離子、子離子、碰撞能量、掃描時間等參數(見表1)。在上述優化條件下,20種農藥均得到較好的分離,混合基質標準工作溶液的總離子流色譜圖見圖1。
2.4基質效應的考察
基質效應是指相同濃度的目標物在純溶劑和基質中的響應存在較大差異,在氣相色譜中主要表現為基質增強效應。這是由于基質成分的存在減少了色譜系統活性位點與待測物分子作用的機會,使得待測物的檢測信號增強。不同目標物、不同基質、不同前處理方法以及不同的儀器狀態下,其基質效應差別較大[10]。消除基質效應影響的方法主要有:加入內標化合物、基質匹配標準工作溶液、加入分析保護劑、應用更多的凈化步驟等。本研究采用基質匹配標準溶液建立標準工作曲線,對基質效應進行補償。
通過對基質標和溶劑標工作曲線斜率的比較,發現采用C18凈化時,只有六氯苯、撲滅津、溴丁酰草胺、環庚草醚、滅藻醌和九氯6種農藥不存在基質效應;而采用QuEChERS前處理方法,克草敵、毒草胺、六氯苯、撲滅津、戊炔草胺、二甲草胺、溴丁酰草胺、環庚草醚、滅藻醌、氯酞酸甲酯、九氯和戊環唑12種農藥均不存在基質效應。實驗結果顯示,采用QuEChERS前處理方法,農藥的基質效應相對較小,上述12種農藥可以采用標樣直接配制工作曲線。
2.5線性范圍、相關系數及定量下限
分別按照乙腈提取-C18柱凈化/GC-MS/MS(方法1)與 QuEChERS/GC-MS/MS(方法2)得到空白大米樣品基質液,制備標準工作曲線溶液,20種農藥的濃度在0.01~0.2 μg/mL范圍內與測得的峰面積呈良好的線性關系,相關系數(r2)均大于0.99,具體數據見表2。分別以3倍和10倍信噪比確定化合物的檢出限(LOD)和定量下限(LOQ),除二丙烯草胺和四溴菊酯的LOQ為10 μg/kg外,其他18種農藥的LOQ均小于5 μg/kg。滿足美國、歐盟、“日本肯定列表”及有關國家對這20種農藥的最高殘留限量(MRLs)要求和方法檢測靈敏度的要求。
2.6回收率與精密度
按照C18柱凈化與QuEChERS凈化對陰性大米樣品進行加標回收實驗,加標水平分別為0.01,0.05,0.20 mg/kg,每個濃度做6次平行,計算平均回收率及相對標準偏差(RSD),結果見表2。從表2數據可知,采用C18柱對大米樣品進行提取凈化,20種農藥的平均回收率為63.3%~96.0%,RSD為1.9%~13.9%;采用QuEChERS法大米樣品進行提取凈化,20種農藥的平均回收率為67.7%~121.0%,RSD為1.9%~16.4%。此兩種方法無明顯差異,均能對批量大米樣品進行同時檢測,且數據準確可靠,回收率與精密度均符合農藥殘留的分析要求。
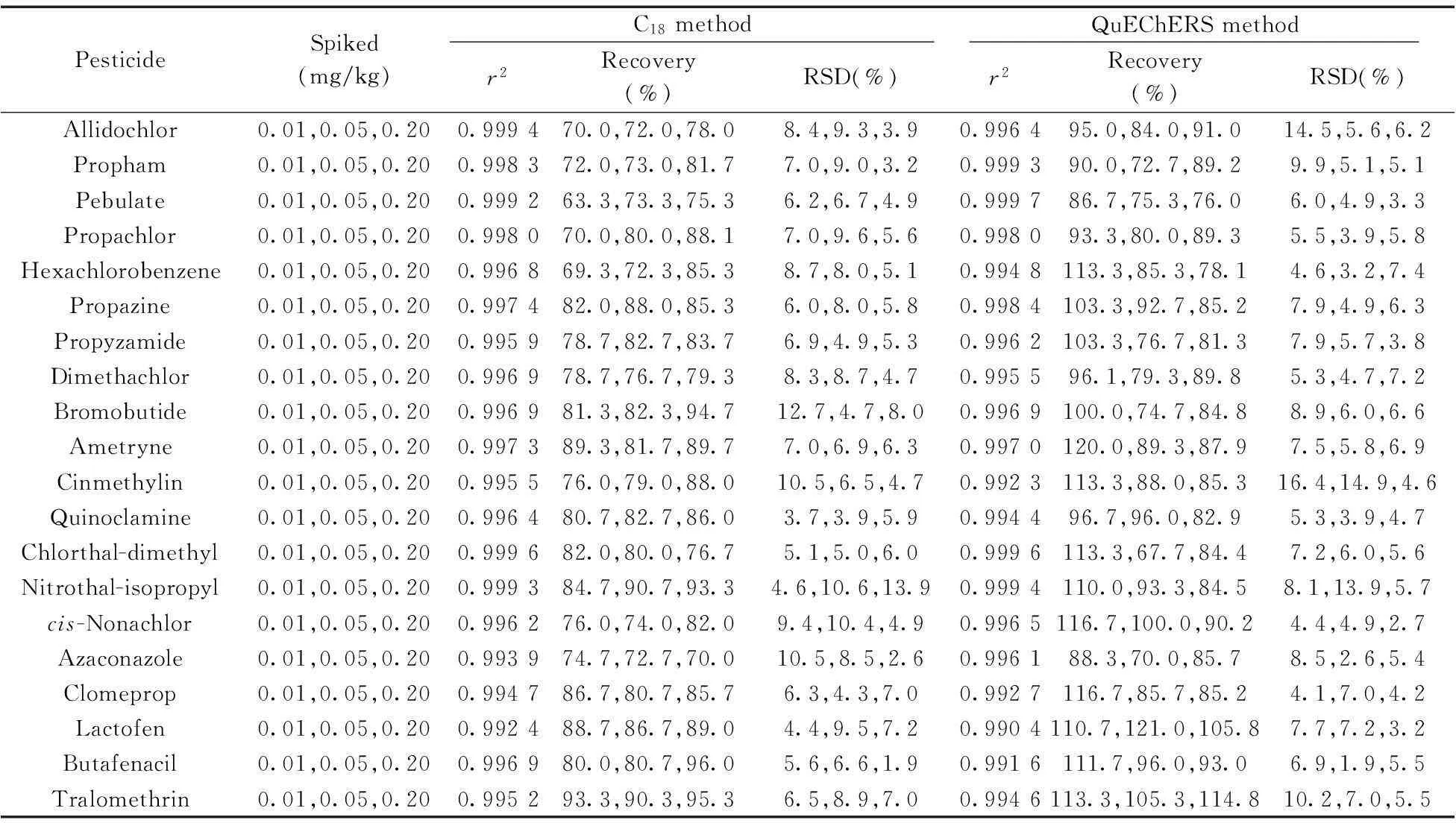
表2 兩種方法的回收率與精密度
2.7大米樣品分析
采用方法1與方法2對市場上購買的3個東北大米、1個盤錦大米、1個泰國大米、1個五常大米樣品中的20種農藥殘留進行分析,每個樣品平行試驗3次,結果均未檢出上述20種農藥。
3結論
本文建立了大米樣品中20種不同種類農藥殘留的兩種GC-MS/MS檢測方法。方法1采用乙腈提取、C18固相萃取凈化、GC-MS/MS分析;方法2采用乙腈提取、QuEChERS法凈化、GC-MS/MS分析。該研究為農藥多殘留檢測提供了不同的思路,方法具有靈敏度高、操作簡單快捷、重現性好且回收率高的特點,適用于大米中多組分農藥殘留的定量檢測與定性確證,在日常實際檢測中取得了滿意的結果。
參考文獻:
[1]Hou X,Han M,Dai X H,Yang X F,Yi S G.FoodChem.,2013,138:1198-1205.
[2]Codex Alimentarius.Pesticide Residues in Food and Feed Codex Pesticides Residues in Food Online Database,2013.http://www.codexalimentarius.net/pestres/data/index.html?lang=en.
[3]European Commission.Pesticides.http://ec.europa.eu/food/plant/pesticides/index_en.htm.
[4]US EPA.Pesticides.http://www.epa.gov/pesticides/.
[5]GB 2763-2014.National Food Safety Standard Maximum Residue Limits for Pesticides in Food.National Standards of the People's Republic of China(食品安全國家標準-食品中農藥最大殘留限量.中華人民共和國國家標準).
[6]Li R,He L,Zhou T,Ji X F,Qian M R,Zhou Y,Wang Q.Anal.Bioanal.Chem.,2014,406:2899-2907.
[7]Liu H J,Guo B Y,Wang H L,Li J Z,Zheng L.Bull.Environ.Contam.Toxicol.,2014,92:451-454.
[8]Paradis D,Bérail G,Bonmatin J M,Belzunces L P.Anal.Bioanal.Chem.,2014,406:621-633.
[9]Anagnostopoulos C,Charalampous A,Balayiannis G.Chromatographia,2015,78:109-118.
[10]Sun Y Y,Sun H,Dong Z L,Gao S J,Du M.FoodRes.Dev.(孫玉玉,孫浩,董振霖,高術杰,杜茂.食品研究與開發),2015,36(3):121-125.
[11]Chen Q Y,Ge B K,Han H F,Wang Y F.J.Instrum.Anal.(陳其勇,葛寶坤,韓紅芳,王云鳳.分析測試學報),2011,30(5):573-576.
[12]Zhang H W,Liu H H,Tian X H,Deng X X,Huang H,Han D F,Xu Y J,Gong X H.J.Chin.MassSpectrom.Soc.(張華威,劉慧慧,田秀慧,鄧旭修,黃會,韓典峰,徐英江,宮向紅.質譜學報),2015,36(2):177-184.
[13]Cao S R,Xu F,Zhang L,Li X L,Wang G M,Ming D W,Yuan R.FoodSci.(曹淑瑞,徐芬,張雷,李賢良,王國民,明德旺,袁若.食品科學),2013,34(12):160-164.
[14]Wang N,Kong D Y,Cai D J,Shi L L,Cao Y C,Pang G F,Yu R B.Environ.Sci.Technol.,2010,44:4334-4340.[15]Li P P,Cheng J,Le Y.J.Instrum.Anal.(李萍萍,程景,樂淵.分析測試學報),2015,34(4):421-427.
[16]Li N,Shi Z H,Pang G F,Fan C L.J.Instrum.Anal.(李南,石志紅,龐國芳,范春林.分析測試學報),2011,30(5):513-521.
[17]Wang X,Wang S J,Cai Z W.TrendsAnal.Chem.,2013,52:170-185.
[18]Ou J F.MatrixEffectsinDeterminationofMultiresiduesofPesticidesinVegetableUsingGasChromatography-MassSpectrometry.Beijing:Chinese of Academy of Agricultural Sciences(歐菊芳.蔬菜中農藥多殘留氣相色譜-質譜法測定中的基質效應研究.北京:中國農業科學院),2008.
[19]Fintschenko Y,Krynitsky A J,Wong J W.J.Agric.FoodChem.,2010,58(10):5859-5861.
[20]Chen N N,Gao H B,Ye N S,Zhong Q D,Xiong Z H,Gu X X.Am.J.Anal.Chem.,2012,3:33-39.
[21]Hernandez F,Cervera M I,Portoles T,Beltran J,Pitarch E.Anal.Methods,2013,5:5875-5984.
[22]Chen X S,Bian Z Y,Yang F,Liu S S,Tang G L,Hu Q Y.Chin.J.Chromatogr.(陳曉水,邊照陽,楊飛,劉珊珊,唐綱嶺,胡清源.色譜),2013,31(11):1116-1128.
[23]Patil S H,Banerjee K,Dasgupta S,Oulkar D P,Patil S B,Jadhav M R.J.Chromatogr.A,2009,1216:2307-2319.[24]Chamkasem N,Ollis L W,Harmon T,Lee S,Mercer G.J.Agric.FoodChem.,2013,61:2315-2329.
[25]Lü B,Chen D W,Miao H.J.Instrum.Anal.(呂冰,陳達煒,苗虹.分析測試學報),2015,34(6):639-645.
[26]Ai L F,Li W,Wang J,Ma Y S,Chen R C,Guo C H.J.Instrum.Anal.(艾連峰,李瑋,王敬,馬育松,陳瑞春,郭春海.分析測試學報),2015,34(5):570-575.
[27]AOAC Official Method 2007.01.Pesticide Residues in Foods by Acetonitrile Extraction and Partitioning with Magnesium Sulfate Gas Chromatography/Mass Spectrometry and Liquid Chromatography/Tandem Mass Spectrometry First Action 2007.AOAC International.
[28]BS EN 15662:2008.Foods of Plant Origin-Determination of Pesticide Residues Using GC-MS and/or LC-MS/MS Following Acetonitrile Extraction/Partitioning and Cleanup by Dispersive SPE-QuEChERS-Method.British Standard.
[29]Anastassiades M,Lehotay S J,Stajnbaher D,Schenck F J.J.AOACInt.,2003,86(2):412-439.
Determination of 20 Kinds of Pesticide Residues in Rice by Gas Chromatography-Tandem Mass Spectrometry
CHEN Xi1,2,DONG Zhen-lin3,SUN Yu-yu3,SHANG Bao-yu2,LIU Jia-cheng2,QU Shi-chao2,JI Ming-shan1*
(1.Plant Protection College,Shenyang Agricultural University,Shenyang110866,China;2.Dalian Entry-Exit Inspection and Quarantine Bureau Technology Center,Zhuanghe116400,China;3.Liaoning Entry-Exit Inspection and Quarantine Bureau Technology Center,Dalian116001,China)
Abstract:Two analytical methods for the simultaneous determination of 20 pesticide residues in rice were proposed.The rice samples were initially extracted with acetonitrile(MeCN), then cleaned up by C18SPE column(method 1) and QuEChERS method(method 2),respectively.The extract was analyzed by GC-MS/MS under selective reaction monitoring(SRM) mode,in which the pesticides were identified by retention time,selected ions and their relative abundances,and quantified by the external standard method.The results indicated that the linear ranges of pesticides were between 0.01 μg/mL and 0.2 μg/mL,with correlation coefficients all above 0.99.At three spiked concentration levels( 0.01,0.05,0.20 mg/kg),the average recoveries of 20 pesticides in rice purified by method 1 ranged from 63.3% to 96.0%,with relative standard deviations(RSDs) of 1.9%-13.9%.The average recoveries of 20 pesticides in rice purified by method 2 ranged from 67.7% to 121.0%,with RSDs of 1.9%-16.4%.The results showed that there was no obvious difference between two pretreatement methods,and this study provided a different pretreatment way for pesticide residue detection.The proposed method was suitable for the simultaneous determination of multiple pesticide residues in rice.
Key words:rice;pesticide residues;gas chromatography-tandem mass spectrometry(GC-MS/MS);QuEChERS;solid phase extraction(SPE)
收稿日期:2015-08-01;修回日期:2015-10-10
基金項目:國家質檢總局科研項目(2010IK134)
*通訊作者:紀明山,博士,教授,研究方向:農藥毒理學,Tel:024-88492673,E-mail:jimingshan@163.com
doi:10.3969/j.issn.1004-4957.2016.04.004
中圖分類號:O657.63;F767.2
文獻標識碼:A
文章編號:1004-4957(2016)04-0394-06
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
當代陜西(2019年8期)2019-05-09 02:22:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
專用汽車(2016年4期)2016-03-01 04:13:43
Coco薇(2015年1期)2015-08-13 02:47:34
