超高效液相色譜-質譜聯用法測定蔬菜中多殺霉素等4種農藥殘留
2016-05-13 09:32:12占繡萍陳建波黃蘭淇余淑紅
分析測試學報 2016年4期
關鍵詞:固相萃取
占繡萍,陳建波*,馬 琳,黃蘭淇,趙 莉,余淑紅
(1.上海市農業技術推廣服務中心,上海 201103;2.上饒市農產品質量安全檢測中心,江西 上饒 333400)
超高效液相色譜-質譜聯用法測定蔬菜中多殺霉素等4種農藥殘留
占繡萍1,陳建波1*,馬琳1,黃蘭淇1,趙莉1,余淑紅2
(1.上海市農業技術推廣服務中心,上海201103;2.上饒市農產品質量安全檢測中心,江西上饒333400)
摘要:采用固相萃取/超高效液相色譜-串聯質譜(SPE/UPLC-MS/MS)建立了多種蔬菜中4種農藥殘留量的分析方法,并對凈化方法、色譜及質譜條件進行優化。蔬菜樣品經乙腈提取、提取液經鹽析后,取上清液經PC/NH2(或NH2)固相萃取小柱凈化,濃縮后,用甲醇定容并過膜,采用超高效液相色譜-串聯質譜多反應監測(MRM)正離子模式進行外標法定量測定。結果表明,4種農藥在5~500 μg/L范圍內線性關系良好(r≥0.999 2),在8種蔬菜中的檢出限和定量下限分別為0.033~0.167 μg/kg和0.10~0.50 μg/kg 。在10,50,250 μg/kg 3個濃度加標水平下,4種農藥的回收率為70.9%~123.4%,相對標準偏差(RSD)小于15%。方法準確、靈敏、簡單、快速,適用于多種蔬菜中多殺霉素等4種農藥殘留的同時測定。
關鍵詞:超高效液相色譜-質譜聯用;固相萃取;農藥殘留;蔬菜
21世紀以來,受氣候、種植制度、栽培方式、生態環境等因素的綜合影響,植物上的病蟲害暴發頻繁,危害程度加重,防控壓力增大,嚴重威脅農業生產安全。特別是隨著設施農業的迅速興起,農作物上的病蟲害不斷加重,許多新的設施病蟲害不斷發生。當下食品安全越來越受到人們重視,高效低毒低殘留農藥的推廣應用必將成為趨勢。多殺霉素、氟硅唑、噻蟲嗪、茚蟲威4種農藥是近年上海市蔬菜作物重點推薦和補貼的農藥新品種,在上海乃至全國范圍內得到了廣泛應用。 為了保證蔬菜質量安全,各國對這些農藥殘留實施了嚴格的監測,我國僅規定了這4種農藥在幾種蔬菜中的限量值,歐盟等發達國家的限量值比較完整,如歐盟規定氟硅唑在所有蔬菜中的限量值均為0.01 mg/kg;多殺霉素的限量值為0.02~10.0 mg/kg;噻蟲嗪的限量值為0.05~10.0 mg/kg;茚蟲威的限量值為0.02~3.0 mg/kg。目前,這些農藥殘留的檢測方法主要包括氣相色譜[1-2]、液相色譜[3-5]、氣相色譜-質譜聯用[6-7]、液相色譜-質譜聯用[8-11]等,但這些方法的適用范圍小,對于復雜基質的農藥殘留測定干擾大,不能同時進行這4種農藥的多殘留檢測。
本文通過反復比較4種SPE柱(不同淋洗液)和3種QuEChERS凈化劑的凈化效果,選擇2種蔬菜對UPLC-MS/MS檢測中的基質效應、提取效率、回收率進行評估研究,優化了空心菜、芹菜、豇豆、黃瓜、番茄、卷心菜、大白菜、絲瓜8種蔬菜中多殺霉素等4種農藥的前處理方法,發現SPE凈化法適合不同品種、大批量樣品的快速檢測,且該方法的檢出限、回收率以及相對標準偏差均能滿足農藥殘留分析要求,能夠較好地為蔬菜中多殺霉素等用量較大的農藥殘留監測及農產品質量控制提供科學依據和技術支持。
1實驗部分
1.1試劑與材料
標準品:多殺霉素、氟硅唑、噻蟲嗪、茚蟲威(純度>99.0%),均為德國Dr.Ehrenstorfer公司提供的有證標準物質。
試劑:氯化鈉(分析純),甲醇、乙腈、甲酸(色譜純),Milli-Q高純水,十八烷基鍵合硅膠(C18)吸附劑(43~60 μm),N-丙基乙二胺(PSA)吸附劑(25~100 μm),石墨化碳黑吸附劑(Carbon SPE bulk sorbent),無水硫酸鎂,石墨碳黑-氨基串聯(PC/NH2)固相萃取柱(500 mg/500 mg/6 mL),氨基(NH2)固相萃取柱(500 mg/6 mL),十八烷基鍵合硅膠(C18)固相萃取柱(500 mg/6 mL),弗羅里硅土(Florisil)固相萃取柱(1 000 mg/6 mL)。凈化小柱和凈化填料由天津博納艾杰爾科技有限公司和美國安捷倫公司提供。
1.2儀器
Agilent1290 UPLC-Agilent 6460 MS超高效液相色譜-三重四極桿串聯質譜聯用儀:配ESI源(美國安捷倫公司);T25DS25高速勻漿分散器(德國IKA公司);GB11240-89電熱恒溫水浴鍋(上海醫療器械五廠);AC-40氮吹濃縮器(安徽黃山醫療器械廠);國華SHAC恒溫振蕩器(常州國華電器有限公司);TDL-5-A離心機(上海安亭科學儀器廠);Milli-Q制水機。
1.3樣品前處理
取不少于1 000 g樣品(空心菜、芹菜、豇豆、黃瓜、番茄、卷心菜、大白菜、絲瓜),去除雜物,取可食部分,將樣品縮分至500 g,經制樣機處理后密封,于4 ℃冷藏保存。
1.3.1樣品提取蔬菜樣品:準確稱取10 g樣品(精確至0.01 g)加入到100 mL離心管中,加入20 mL乙腈,高速勻漿1 min,以5 000 r/min離心5 min,上清液轉移至加入3~5 g 氯化鈉的50 mL離心管中,劇烈振搖1 min,室溫下靜置1 h。
1.3.2樣品凈化空心菜、芹菜、豇豆、黃瓜(色素較深)樣品:移取上清液5 mL至50 mL 小燒杯中,在50 ℃水浴溫度下氮吹至約 1 mL。上樣前,先用5 mL乙腈-甲苯(3∶1,體積比)預淋PC/NH2串聯柱,棄去;當液面到達填料頂部時,迅速將樣品濃縮液轉移至凈化柱上,再用5 mL乙腈-甲苯(3∶1)洗滌樣液燒杯,并將洗滌液轉移至凈化柱中,再重復4次洗滌轉移,收集上述25 mL洗脫液于100 mL燒杯中,50 ℃水浴溫度下氮吹至干。加入2.5 mL 乙腈混勻定容,經0.22 μm微孔有機濾膜過濾后,供超高效液相色譜-質譜測定。
番茄、卷心菜、大白菜、絲瓜(色素較淺)樣品:用NH2柱替代PC/NH2串聯柱,其他相同。
1.4色譜-質譜條件
1.4.1色譜條件Poroshell 120 EC-C18反相液相色譜柱(3.0 mm×100 mm,2.7 μm);流動相:A為5 mmol/L乙酸銨水溶液,B為乙腈;流速0.40 mL/min;柱溫40 ℃;進樣量2 μL 。梯度洗脫程序:0~0.5 min,95% A;0.5~4.0 min,95%~20% A;4.0~7.0 min,20%~5% A。停止時間7.0 min 。
1.4.2質譜條件離子源:大氣壓電噴霧電離,正離子模式(ESI+);毛細管電壓:3.5 kV;離子源干燥氣溫度:300 ℃;鞘氣溫度:350 ℃;鞘氣流量:12 L/min;掃描方式:多反應監測(MRM);5.5 min后加電子倍增管電壓(EMV) 300 V。4種農藥的質譜分析參數見表1。

表1 4種農藥的多反應監測質譜分析參數
*quantitative ion
2結果與討論
2.1樣品前處理條件的優化
2.1.1凈化條件的優化蔬菜樣品基質中含有較多的有機酸、維生素以及脂肪等,這些物質帶來的基質效應將會嚴重影響檢測結果的準確性[12]。為降低或消除基質效應,選擇合適的樣品前處理方法至關重要[13]。本研究對固相萃取(SPE) 與基質固相分散萃取法兩種凈化技術進行了比較。
采用SPE柱進行凈化時,選擇了NH2,Florisil,C18,PC/NH24種固相萃取柱,并選擇不同的淋洗液進行洗脫。NH2-SPE:二氯甲烷+甲醇(95+5) 4 mL預淋,10 mL洗脫;Florisil-SPE:丙酮+正己烷(10+90) 4 mL預淋,10 mL洗脫;C18-SPE:乙腈+甲苯(3+1) 5 mL預淋,10 mL洗脫;PC/NH2-SPE:乙腈+甲苯(3+1) 5 mL預淋,10 mL洗脫,收集第1瓶(石墨化碳易吸附農藥,所以洗脫液又增加15 mL,收集第2瓶);分別收集洗脫液,吹干,定容,待測。
采用基質固相分散萃取(類似QuEChERS)凈化法時,選擇3種不同的凈化填料進行篩選:QuE1(0.1 g PSA-0.3 g MgSO4);QuE2(0.1 g PSA-0.3 g MgSO4-0.05 g Carb);QuE3(0.3 g MgSO4-0.1 g C18-0.1 g PSA)。其他步驟參照文獻[14]。
以基質較復雜、色素較深的空心菜為例,添加100 μg/kg的混標至空心菜空白樣品中,重復測定3次,以純標計算得到的回收率結果如表2所示。
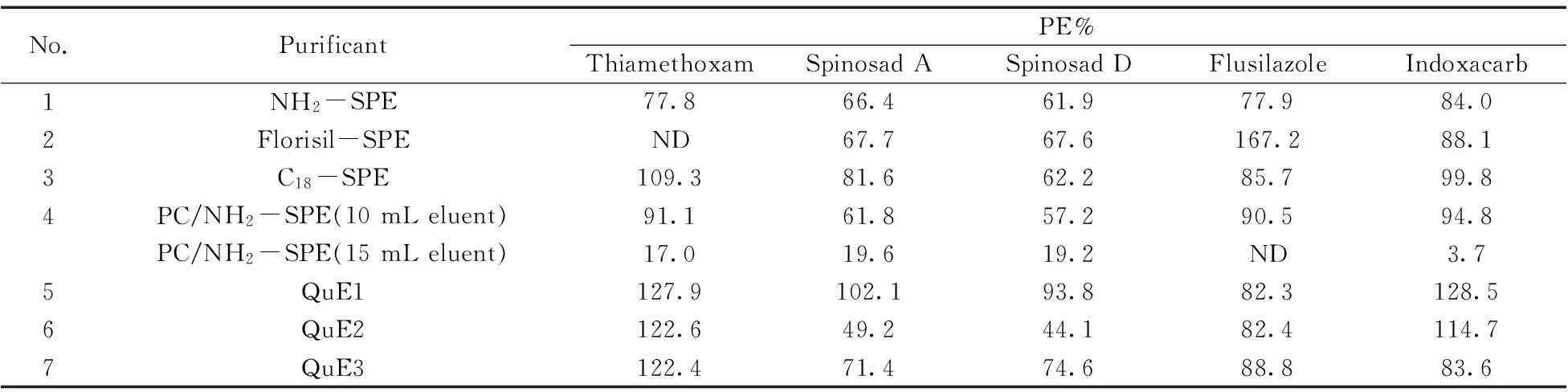
表2 不同凈化條件下空心菜樣品加標回收率的比較
ND:no detected
通過以上7種方法的比較,并綜合去除色素效果和回收率結果,發現方法4(淋洗液10+15 mL)既能保證方法回收率,又能除去色素等雜質,有利于保護色譜柱、離子源等,延長色譜柱、儀器的壽命。
根據上述試驗,發現淋洗液用乙腈-甲苯混合溶劑,空心菜樣品中5種化合物的回收率和雜質的去除效果最佳。
實驗繼續考察了色素較淺的番茄空白樣添加100 μg/kg標樣后的回收率,重復3次,并篩選了PC/NH2,NH2,C18,QuE3 4種凈化方法,SPE的洗脫液統一用乙腈-甲苯(3∶1,體積比)25 mL,QuE3(0.3 g MgSO4-0.1 g C18-0.1 g PSA),實驗結果見表3 。
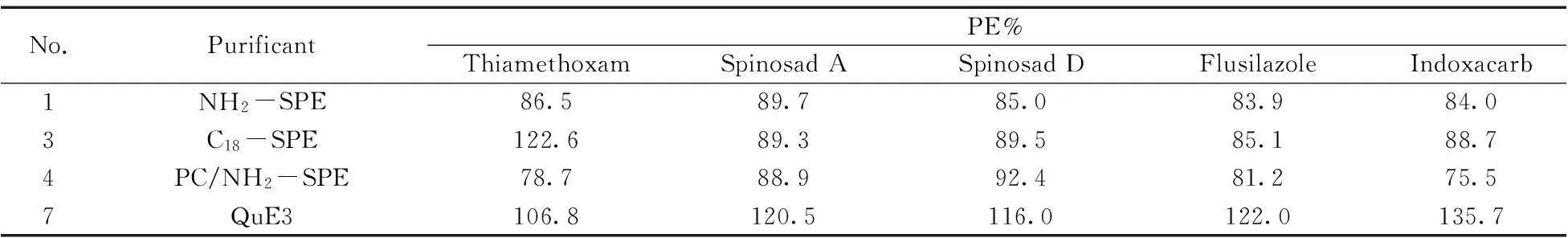
表3 不同凈化方法在番茄空白樣品中加標回收率的比較
從表3結果可知,QuE3法的回收率偏高,去除雜質效果有限,基質效應較明顯;C18-SPE柱中噻蟲嗪的回收率偏高,NH2柱和PC/NH2串聯柱的回收率基本能滿足要求,PC/NH2串聯柱的回收率比NH2柱的回收率低,可能是由于PC/NH2串聯柱中的PC較容易吸附農藥。NH2柱的價格低,故從成本和回收率結果考慮,對于色素較低的樣品采用NH2柱凈化。
2.1.2SPE方法中淋洗液體積的優化選擇PC/NH2-SPE,乙腈-甲苯(3∶1)混合溶劑,考察了不同淋洗液體積(10,15,20,25,30 mL)對空心菜中噻蟲嗪、多殺霉素A、多殺霉素D、氟硅唑、茚蟲威回收率(加標100 μg/kg)的影響。結果表明,隨著乙腈-甲苯淋洗劑體積的增大,噻蟲嗪、多殺霉素A、多殺霉素D的回收率增加,氟硅唑、茚蟲威的回收率增加不明顯。當淋洗液體積達到25 mL時回收率基本能滿足分析要求;當洗脫液在30 mL時回收率較高,但噻蟲嗪的回收率偏高是由于雜質干擾增強。所以確定淋洗劑體積為25 mL。
選擇NH2柱,乙腈-甲苯(3∶1)混合溶劑,考察了不同淋洗體積(10,15,20,25,30 mL)對番茄中噻蟲嗪、多殺霉素A、多殺霉素D、氟硅唑、茚蟲威回收率(100 μg/kg)的影響。結果表明,氟硅唑、茚蟲威的回收率在10 mL洗脫液時已能滿足要求;噻蟲嗪、多殺霉素A、多殺霉素D的回收率隨著淋洗液的增加而增加,當淋洗液體積達到25 mL時洗脫液較為澄清,且回收率基本能滿足分析要求;當洗脫液在30 mL時回收率較高,但洗脫液的顏色加深且渾濁,噻蟲嗪的回收率明顯偏高,雜質干擾增強。因此,確定最佳淋洗劑體積為25mL。
2.2色譜條件的選擇
2.2.1流動相的篩選比較了甲醇-水、乙腈-水、甲醇-0.1%甲酸水、乙腈-0.1%甲酸水流動相體系的分離效果,由于4種農藥均為正離子電離模式,在流動相中加入甲酸后4種農藥更易于電離,且靈敏度更高,所以選擇水相中加一定的甲酸。有機溶劑相使用甲醇時,多殺霉素D和氟硅唑很難分開;而用乙腈則可以得到較好地分離,所以最終選擇乙腈-0.1%甲酸水為流動相。
2.2.2色譜柱比較了常用的3種超高效液相色譜柱:poroshell 120 EC-C18(3.0 mm×100 mm,2.7 μm),poroshell 120 EC-C18(3.0 mm×50 mm,2.7 μm),ZORBAX Eclipse Plus C18(2.1 mm×50 mm,1.8 μm)。發現100 mm長柱比50 mm短柱能更好地分離以上4種農藥,所以選擇poroshell 120 EC-C18(3.0 mm×100 mm,2.7 μm)進行分離。
2.2.3柱溫考察了柱溫(20,30,40 ℃)對分離效果的影響,發現4種農藥的靈敏度和穩定性均變化不明顯。但柱溫高時,泵壓力較小,樣品溶液中的顆粒不易凝固在柱中,所以選擇溫度40 ℃,以保證樣品溶液順利通過色譜柱,較好的延長色譜柱壽命。
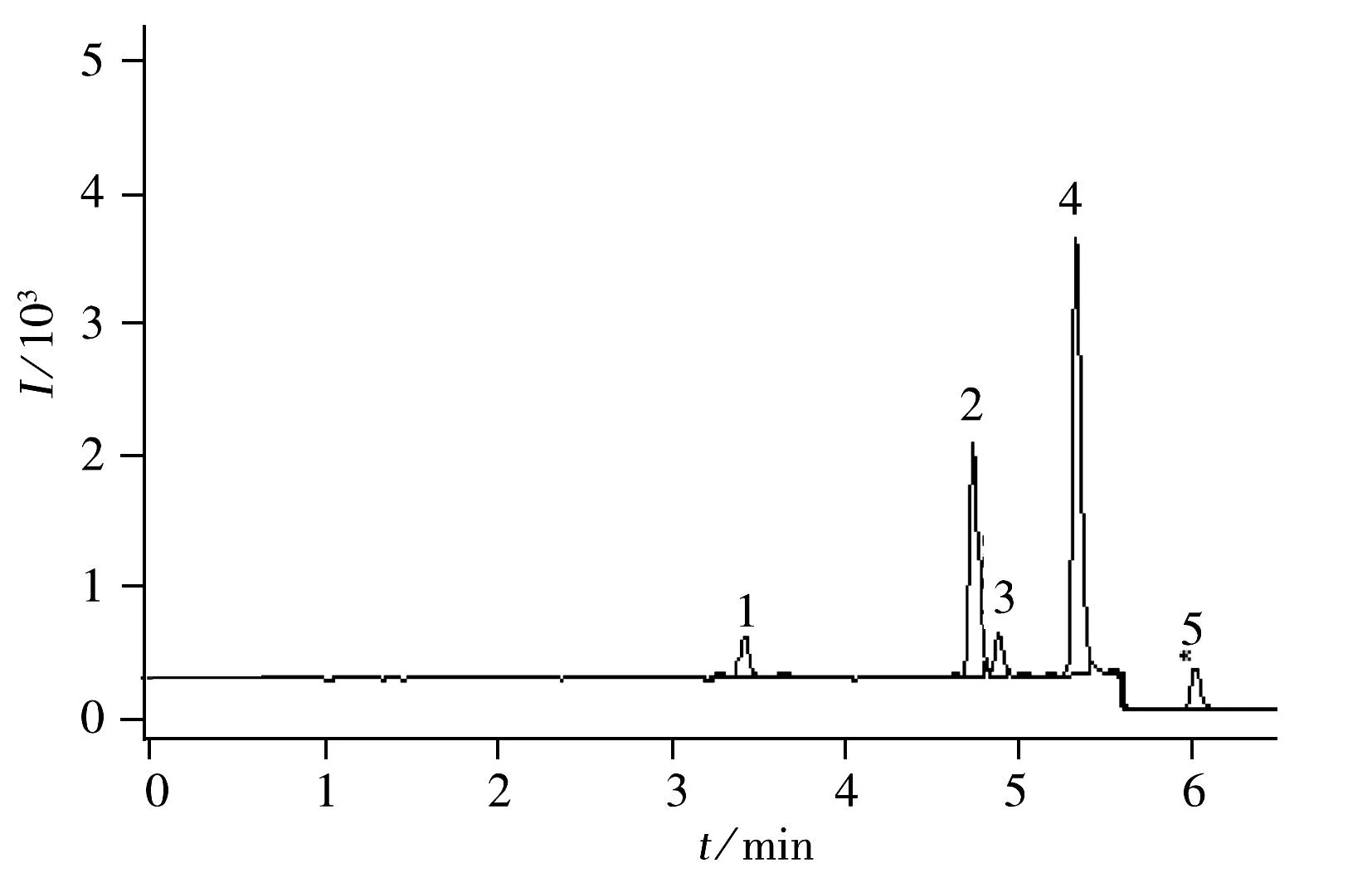
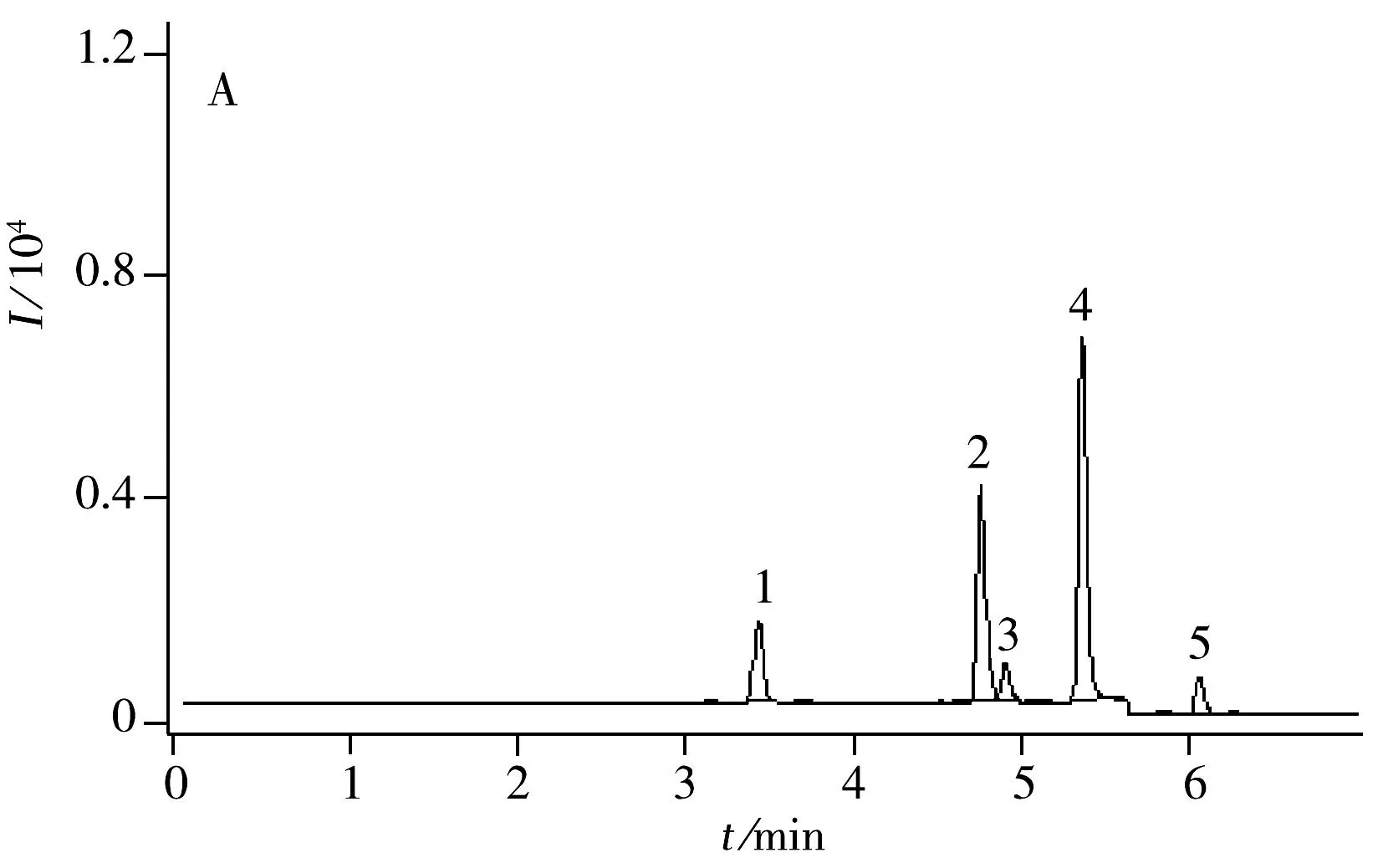
圖1 4種農藥標準溶液(5 μg/L)在MRM模式下的UPLC-MS/MS色譜圖Fig.1 UPLC-MS/MS chromatogram of 4 pesticides(5 μg/L)1.thiamethoxam,2.spinosad A,3.spinosad D,4.flusilazole,5.indoxacarb
2.3質譜條件的選擇
將4種農藥標準儲備溶液稀釋至500 μg/L,分別進行一級質譜掃描以確定各農藥的準分子離子峰;再分別以其準分子離子進行選擇離子監測(MS2SIM),并優化碎裂電壓(Fragment);再通過子離子掃描(Product ion scan)確定定量離子和定性離子,最后采用多反應監測(MRM)采集模式進行碰撞能量的進一步優化。因為茚蟲威的靈敏度較低,所以在茚蟲威出峰(5.5 min)時開始加電壓(Delta EMV)300 V,可以更好地提高其響應值。
2.4檢出限與線性范圍
配制4種農藥的混合標準溶液,質量濃度分別為5,10,50,100,250,500 μg/L,進樣2 μL,以MRM定量離子的色譜峰面積(Y)對分析物的質量濃度(X,μg/L)繪制標準曲線,以3倍信噪比(S/N)確定檢出限(LOD),10倍信噪比(S/N)確定定量下限(LOQ),結果見表4 。4種農藥在5~500 μg/L范圍內線性關系良好,LOD為0.033~0.167 μg/kg,LOQ為0.10~0.50 μg/kg。圖1為5 μg/L標準品的MRM色譜圖。
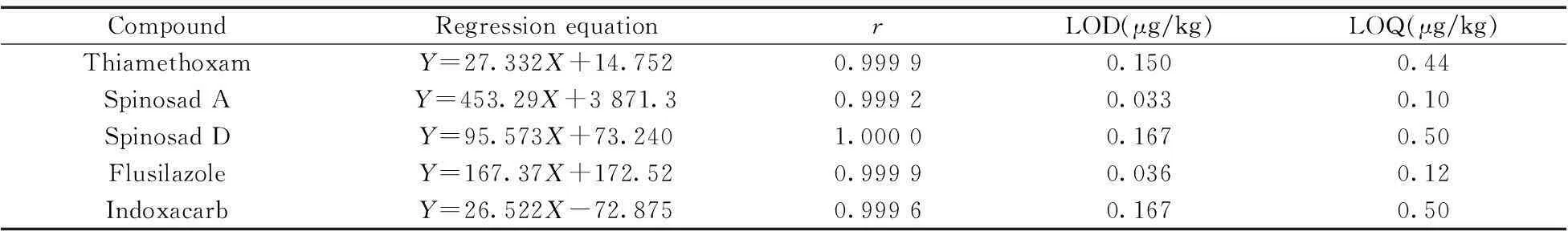
表4 4種農藥的線性方程、相關系數(r)、 檢出限(LOD)和定量下限(LOQ)
2.5回收率與精密度
分別采用空心菜、芹菜、豇豆、黃瓜、番茄、卷心菜、大白菜、絲瓜8種蔬菜進行添加回收實驗和精密度實驗,分別以10,50,250 μg/kg 作為3個添加水平,每個添加水平做5次平行實驗,結果如表5所示。
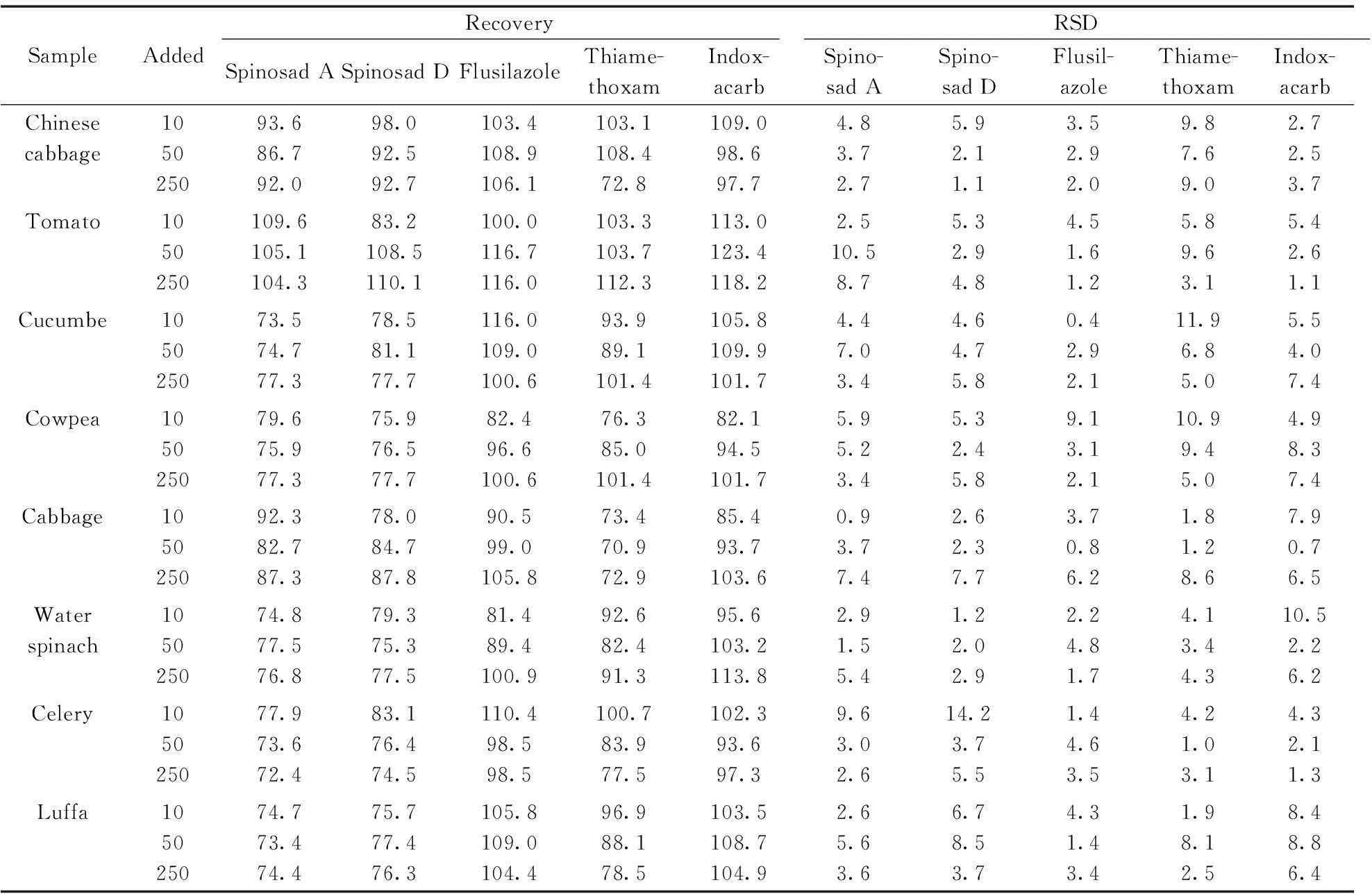
表5 3個加標水平下農藥的回收率及精密度(n=5)
4種農藥(5種化合物)的平均回收率為70.9%~123.4%,相對標準偏差(RSD)為0.7%~14.2 %,表明該方法有良好的精密度和準確度,能達到殘留分析的要求。圖2為空心菜和番茄添加10 μg/kg樣品的UPLC-MS/MS色譜圖。
2.6蔬菜樣品的檢測
應用本方法對市售多種蔬菜共100個樣品進行隨機抽檢。在杭白菜、空心菜、米莧、芹菜、青菜、生菜等12個樣品中檢出茚蟲威(殘留量0.01~0.25 mg/kg),茼蒿、莧菜、芹菜3個樣品中檢出噻蟲嗪(殘留量0.01~0.16 mg/kg),莧菜等2個蔬菜樣品檢出氟硅唑(殘留量0.03~0.04 mg/kg)。查詢國標、CODEX/FAO、歐盟、日本等對噻蟲嗪等4種農藥在相應蔬菜上最大殘留限量值(MRL),發現參照歐盟標準:氟硅唑在所有蔬菜中的MRL為0.01 mg/kg(國標暫未規定氟硅唑在莧菜中的MRL,規定其在玉米筍中為0.01 mg/kg,刀豆、番茄0.1 mg/kg,黃瓜1 mg/kg)。本次抽檢的100個蔬菜樣品中,有2個莧菜樣品超標(氟硅唑參照歐盟蔬菜上的殘留限量值),需謹慎使用。
參考文獻:
[1]Zhu L P,Dong J,Sun J,Pan S Q.Chin.J.Anal.Lab.(朱麗萍,董靜,孫軍,潘守奇.分析試驗室) ,2008,27(8):79-82.
[2]Lu S M,Wang X C,Zhang J T,Ma Y E.Mod.Agrochem.(陸松茂,汪學才,張俊濤,馬又娥.現代農藥),2007,6(5):35-38.
[3]Jian Q,Zhu G Y,Zheng Z T.Agrochemicals(簡秋,朱光艷,鄭尊濤.農藥),2015,54(1):51-52.
[4]Chen L,Chen J M,Xia F L,Dai L C,Yu P Z.Chin.J.Pestic.(陳莉,陳家梅,夏福利,戴榮彩,余蘋中.農藥學學報),2004,6(2):87-89.
[5]Liu B,Guo D L,Mao J S,Zhao S C,Wang Y T.Agrochemcals(劉賓,郭棟梁,毛江勝,趙善倉,王玉濤.農藥),2009,48(9):667-668,674.
[6]Wang L Z,Wang D F,Zheng J C,Wang R L,Liu Y N.Phys.Test.Chem.Anal.:Chem.Anal.(王連珠,王登飛,鄭俊超,王瑞龍,劉溢娜.理化檢驗:化學分冊) ,2006,42(12):1025-1029.
[7]Zhang F,Zhang X Z,Luo F J,Chen Z M,Sun W J,Liu G M,Lou Z Y.J.Instrum.Anal.(張芬,張新忠,羅逢健,陳宗懋,孫威江,劉光明,樓正云.分析測試學報),2013,32(4):393-400.
[8]GB/T 20769-2008.Determination of 450 Pesticides and Relates Chemicals Residues in Fruits and Vegetables—LC-MS-MS Method(水果和蔬菜中450種農藥及相關化學品殘留量的測定 液相色譜-串聯質譜法.中華人民共和國標準).
[9]Sun M,Cao Z Y,Liu H,Ma Y N,Chen M X.Chin.J.Anal.Lab.(孫敏,曹趙云,劉慧,馬有寧,陳銘學.分析試驗室) ,2010,29(8):70-74.
[10]Wu Y,Jiang B,Xu Y G,Zhao W,Meng X R,Zhou Y,Yu J H,Zu Y G.Chin.J.Chromatogr.(吳巖,姜冰,徐義剛,趙偉,孟祥瑞,周原,于佳會,祖元剛.色譜) ,2015,33(3):228-234.
[11]Jiang B W.FoodSci.(江濱煒.食品科學),2009,30(10):176-178.
[12]Zhou L,Wang X Q,Xu H,Wang X Y,Liu X C,Liu Z W,Liu Q J,Zhang H.Phys.Test.Chem.Anal.:Chem.Anal.(周莉,王新全,徐浩,王祥云,劉芯成,劉之偉,劉秋菊,章虎.理化檢驗:化學分冊),2009,28(6):753-756.[13]Xiang P,Shen M,Zhuo X Y.J.Instrum.Anal.(向平,沈敏,卓先義.分析測試學報),2009,28(6):753-756.[14]Yi X B,Liang Y S,Huang X Q,Sun H Z,Jiang C,Liu S Q.J.Instrum.Anal.(易錫斌,梁玉樹,黃曉琴,孫慧珍,江楚,劉世琦.分析測試學報),2015,34(7):829-835.
Simultaneous Determination of Four Pesticides Residues in Vegetables by Ultra Performance Liquid Chromatography-Tandem Mass Spectrometry
ZHAN Xiu-ping1,CHEN Jian-bo1*,MA Lin1,HUANG Lan-qi1,ZHAO Li1,YU Shu-hong2
(1.Shanghai Agriculture Technical Extension Service Center,Shanghai201103,China;2.Shangrao Agricultural Product Quality and Safety Testing Center in Jiangxi Province,Shangrao333400,China)
Abstract:A method was developed for the determination of four pesticides residues in a variety of vegetables using solid-phase extraction(SPE) and ultra performance liquid chromatography-triple quadrupole-mass spectrometry(UPLC-MS/MS).The purification methods(solid phase extraction column,QuEChERS method,elution solvent,elution volume),chromatographic conditions(mobile phase type,flow rate and column temperature) and mass spectrometric parameters(fragment,delta EMV) were optimized.The pesticides samples were extracted with acetonitrile,and then cleaned up on a Carbon/NH2(or NH2) SPE column.The eluting solution was evaporated and made up to a definite volumn with methanol,finally analyzed by reversed-phase ultra performance liquid chromatography under multiple reaction monitoring(MRM) of positive ion mode with the external standard method.The calibration curves of four pesticides showed good linearities in the range of 5-500 μg/L with correlation coefficients not less than 0.999 2.The limits of detection(LOD) were in the range of 0.033-0.167 μg/kg and the limits of quantitation(LOQ) were in the range of 0.10-0.50 μg/kg for 4 pesticides spiked in vegetable.The recoveries of 4 pesticides in vegetable sample spiked with three concentration levels of 10,50,250 μg/kg ranged from 70.9% to 123.4% with RSDs less than 15%.The established method was accurate,sensitive and simple,and was suitable for the simultaneous analysis of various pesticides residues.
Key words:ultra performance liquid chromatography-tandem mass spectrometry(UPLC-MS/MS);solid-phase extraction(SPE);pesticide residue;vegetable
收稿日期:2015-09-30;修回日期:2015-11-07
基金項目:上海市農業委員會青年科技基金(滬農青字(2014)第4-11號)
*通訊作者:陳建波,碩士,農藝師,研究方向:農藥質量與殘留分析,Tel:021-64059431,E-mail:cjb0123@126.com
doi:10.3969/j.issn.1004-4957.2016.04.018
中圖分類號:O657.63;F767.2
文獻標識碼:A
文章編號:1004-4957(2016)04-0476-06
猜你喜歡
分析化學(2016年7期)2016-12-08 00:54:07
中國科技博覽(2016年2期)2016-04-25 14:11:43
湖北工業職業技術學院學報(2016年1期)2016-04-20 17:12:54
分析化學(2015年10期)2015-11-03 07:52:24
食品安全導刊(2015年10期)2015-10-26 04:44:22
安徽農學通報(2015年18期)2015-10-20 00:50:11
安徽農學通報(2015年17期)2015-09-30 00:52:24
分析化學(2015年9期)2015-09-11 07:09:54
肉類研究(2015年5期)2015-08-08 12:46:08
肉類研究(2015年3期)2015-06-16 12:40:36
