超高效液相色譜-串聯質譜法同時測定紡織品中4種苯并三唑類紫外穩定劑
2016-05-13 09:32:00施點望薛建平
分析測試學報 2016年4期
關鍵詞:紡織品
朱 峰,衛 敏,施點望,薛建平
(1.福建省纖維檢驗局,福建 福州 350026;2.福建省紡織產品檢測技術重點實驗室,福建 福州 350026)
超高效液相色譜-串聯質譜法同時測定紡織品中4種苯并三唑類紫外穩定劑
朱峰1,2*,衛敏1,2,施點望1,2,薛建平1,2
(1.福建省纖維檢驗局,福建福州350026;2.福建省紡織產品檢測技術重點實驗室,福建福州350026)
摘要:建立了快速同時測定紡織品中UV-350,UV-320,UV-328和UV-327 4種苯并三唑類紫外穩定劑(BUVSs)的超聲提取/超高效液相色譜-串聯質譜(USE-UPLC-MS/MS)分析方法。樣品經正己烷飽和的乙腈超聲波提取后,采用Eclipse Plus C18柱(2.1 mm×50 mm,1.8 μm)分離,甲醇-0.01 mol/L甲酸銨水溶液為流動相梯度洗脫,電噴霧離子源電離,正離子多反應監測模式進行定性和定量分析,外標法定量。結果表明,4種BUVSs的線性相關系數(r2)均大于0.99,檢出限為0.006 3~0.024 mg/kg,定量下限為0.021~0.081 mg/kg。加標回收率為80.0%~102.9%,相對標準偏差(RSD)為1.2%~9.9%。將該方法應用于紡織品中BUVSs的分析測定,結果令人滿意。
關鍵詞:超高效液相色譜-串聯質譜;苯并三唑類紫外穩定劑;紡織品;超聲提取
苯并三唑類紫外穩定劑(Benzotriazole UV stabilizers,BUVSs)作為一種紫外吸收劑,已被廣泛用于各種工業產品和消費產品[1],如建筑材料、汽車組件、油漆、鞋、化妝品、織物、高分子材料等領域[2-3]。經過BUVSs處理的紡織品,不僅可以保護人體皮膚免受過多紫外線的傷害,還可避免紫外線催化反應破壞高分子結構,提高織物上染料、涂料和纖維等的防曬、耐候、抗老化能力[4]。但已有研究表明部分BUVSs為具有持久性、生物蓄積性和毒性的化合物[5],可能破壞生物體的內分泌系統,對生物體的生殖和發育產生不利影響[6-7],并可能對人類有致癌作用[8]。早在1970年,Jungclaus等[9]就在污水、河水以及沉積物中發現了2-(3,5-二叔丁基-2-羥基苯)苯并三唑(UV-320)、2-(3,5-二叔丁基-2-羥苯基)-5-氯苯并三唑(UV-327)、2-(3,5-二叔戊基-2-羥苯基)苯并三唑(UV-328)。2012年,Ruan等[10]首次在中國污水污泥中檢出2-(3-仲丁基-5-叔丁基-2-羥苯基)苯并三唑(UV-350)等苯并三唑類化合物。2014年12月,歐洲化學品管理局(ECHA)將UV-320,UV-328列入第十二批SVHC清單,并將其歸為持久、生物累積、有毒物質(PBT)。2015年8月,ECHA公布包括UV-327,UV-350在內的7種潛在SVHC咨詢清單,展開公眾評議。因此,建立紡織品中4種BUVSs的同時檢測方法,對于應對貿易壁壘、提升紡織品質量技術水平和污染控制具有重要意義。
BUVSs的檢測方法主要有氣相色譜-質譜法(GC-MS)[1,11-12]、氣相色譜-串聯質譜法(GC-MS/MS)[13-14]、液相色譜-串聯質譜法(LC-MS/MS)[10,15-19],研究基質主要集中在沉積物[9-10,13]、海洋哺乳動物[1]、水體[11,18-19]、土壤[12]、塵埃[14,17]、魚類[15-16]等生物和環境樣本。超高效液相色譜-串聯質譜(UPLC-MS/MS)技術因具有靈敏度高、檢出限低的特點,可以進行多組分定性、定量檢測,是目前報道較多的方法。目前國內尚無相關的國家或行業標準,紡織品中4種BUVSs組分的同時測定也未見文獻報道。
本文建立了同時檢測紡織品中4種苯并三唑類紫外穩定劑(見表1)的超聲提取/超高效液相色譜-串聯質譜(USE/UPLC-MS/MS)分析方法。方法簡單、快速、靈敏、準確,并用于紡織樣品中4種BUVSs的檢測,為生態紡織品的檢測及監控提供了科學依據和技術支持,為制訂相關檢測標準提供技術支撐和參考。

表1 4種BUVSs的化學信息
1實驗部分
1.1儀器與試劑
1290 Infinity超高效液相色譜儀,6410B三重四極桿質譜儀,配電噴霧離子源(ESI)及MassHunter Workstation 數據處理系統(美國Agilent公司);97043-942超聲波發生器(美國VWR公司)。
標準對照品:UV-350(97.0%,Tokyo Chemical Industry);UV-320(99.0%,德國Dr.Ehrenstorfer公司);UV-328、UV-327(98.0%,Tokyo Chemical Industry);甲醇、正己烷、乙腈(色譜純,德國CNW科技公司);甲酸銨(98.0%,德國CNW科技公司);實驗用水為超純水。
1.2標準溶液的配制
準確稱取適量的標準品,用乙腈溶解,配成質量濃度為200 mg/L的標準儲備液,于4 ℃下保存。使用時,用正己烷飽和的乙腈逐級稀釋至所需濃度。
1.3樣品前處理
試樣剪碎至5 mm×5 mm,混勻后,準確稱取1.0 g樣品(精確至0.000 1 g),置于25 mL具塞錐形瓶中,加入15 mL正己烷飽和的乙腈,于60 ℃超聲波發生器中提取15 min。將提取液過濾,殘渣再用15 mL正己烷飽和的乙腈超聲提取15 min,合并濾液于25 mL容量瓶中,以正己烷飽和的乙腈定容,經0.22 μm有機濾膜過濾,用UPLC-MS/MS檢測。
1.4色譜及質譜條件
1.4.1UPLC條件色譜柱:Agilent Zorbax Eclipse Plus C18柱(2.1 mm×50 mm,1.8 μm;美國Agilent公司);流動相:A為甲醇;B為0.01 mol/L甲酸銨水溶液。洗脫梯度:0~7 min,88%~90% A;7~8 min,90%~100% A;8~10 min,100% A;10~10.5 min,100%~88% A;10.5~12 min,88% A。流速:0.3 mL/min;柱溫:20 ℃;進樣體積:5.0 μL。
1.4.2MS/MS條件離子源:ESI;掃描方式:正離子掃描;檢測方式:多反應監測(MRM)模式;毛細管電壓:4 000 V;霧化氣(氮氣)壓力:2.41×105Pa;干燥氣(氮氣)溫度:350 ℃;干燥氣流速:12 L/min;其他實驗參數見表2。

表2 多反應監測模式下4種BUVSs的串聯質譜參數及保留時間
*quantitative ion
2結果與討論
2.1色譜柱的選擇
4種BUVSs分子量接近,結構類似,分離難度較大,而UV-350和UV-320為同分異構體,因此兩者的分離是色譜-質譜檢測的關鍵。實驗考察了Zorbax SB-C18柱(2.1 mm×150 mm,3.5 μm)、Poroshell 120 Phenyl Hexyl柱(2.1 mm×100 mm,2.7 μm)、Poroshell 120 SB-C18柱(2.1 mm×100 mm,2.7 μm)和Zorbax Eclipse Plus C18柱(2.1 mm×50 mm,1.8 μm)對4種BUVSs的保留和分離效果,發現在同等條件下,Zorbax SB-C18柱、Poroshell 120 Phenyl Hexyl柱的分離效果較差,Poroshell 120 SB-C18和Zorbax Eclipse Plus C18柱對4種BUVSs尤其是UV-350和UV-320可實現基線分離,均可采用。由于4種BUVSs在Zorbax Eclipse Plus C18柱上的保留時間更短,分離效率更高,因此實驗選擇Zorbax Eclipse Plus C18柱(2.1 mm×50 mm,1.8 μm)作為分離柱。
2.2流動相的選擇
為了實現目標化合物間良好的色譜分離及響應,分別考察了甲醇-水溶液和乙腈-水溶液流動相體系對4種BUVSs分離的影響。結果表明:甲醇流動相體系和乙腈流動相體系均能較好地實現4種BUVSs中同分異構體的分離,但甲醇流動相體系下目標化合物的響應顯著大于乙腈流動相體系。為了進一步改善質譜信號響應和分離效果,分別在水相中添加0.1%甲酸、0.01 mol/L甲酸銨、0.01 mol/L甲酸銨+0.1%甲酸,發現水相中添加0.01 mol/L甲酸銨水溶液時,色譜峰的離子化效果最好、響應最高,因此實驗選擇甲醇-0.01 mol/L甲酸銨水溶液為流動相。
為進一步改善同分異構體間的分離度,選擇梯度洗脫方式,優化梯度條件,并對柱溫和流速進行了優化,實現了UV-350和UV-320同分異構體的基線分離。按“1.4.1”所述UPLC條件設置,分離效果最佳、響應最高。
2.3質譜條件的選擇
采用三通閥自動進樣方式優化質譜條件,對5 mg/L單一目標化合物的乙腈溶液進行ESI正、負離子全掃描檢測。結果表明,各紫外穩定劑在負離子模式下的質譜響應較弱,而在正離子模式下的分子離子峰響應較高,因此選擇在ESI正離子掃描模式下對4種BUVSs進行分析。
在該模式下,所有分析物經ESI電離生成特征離子峰[M+H]+,將其選為母離子峰。對選定的母離子進行子離子掃描,選取豐度較強且干擾較少的子離子用于定性定量計算,通過多反應監測掃描(MRM)對碰撞能量、碎裂電壓、儀器干燥氣溫度、干燥氣流速、霧化氣壓力以及毛細管電壓等參數進行優化,使特征碎片的離子強度達到最大,確定最佳的質譜參數如“1.4.2”所述。
圖1為4種BUVSs的LC-MS/MS多反應監測色譜圖。



圖1 4種BUVSs的MRM色譜圖Fig.1 Chromatograms of 4 BUVSs in MRM modepeak numbers denoted are the same as those in Table 2
2.4前處理方法的優化
超聲波萃取常用于紡織品中有毒有害物質的提取,本文對影響超聲波萃取效率的主要因素(提取溶劑種類、提取溫度、提取時間、提取溶劑體積)進行了考察。
由于苯并三唑類化合物溶于乙腈、甲醇、二氯甲烷等有機溶劑,因此考察了乙腈、甲醇、正己烷、二氯甲烷、乙酸乙酯5種溶劑的提取效果。稱取1.0 g(精確至0.000 1 g)代表性自制紡織品陽性樣品,分別加入15 mL提取溶劑,在30 ℃下超聲提取30 min,收集濾液于50 mL濃縮瓶中,于40 ℃水浴旋轉蒸發器濃縮至近干,用乙腈溶解并定容至2.0 mL,經有機濾膜過濾后進樣分析,平行測定3個樣品,計算4種BUVSs的回收率。結果表明:乙腈對4種BUVSs的萃取回收率最佳,均高于90%,且乙腈和正己烷作為提取溶劑時回收率最為穩定,其標準偏差均小于3.5%。為改善提取溶劑的極性和對4種BUVSs萃取的選擇性,進一步考察了正己烷飽和的乙腈為提取溶劑時的效果,發現4種BUVSs的回收率和穩定性均較好,且ESI全掃描結果顯示其提取液基底干擾較少。因此,實驗選擇正己烷飽和的乙腈作為提取溶劑。
進一步優化提取溫度、提取時間、提取溶劑體積等參數。結果顯示:隨著提取溫度從常溫升至60 ℃、提取時間從10 min增至30 min、提取溶劑體積從10 mL增至30 mL,萃取回收率逐漸增加,此后繼續增加上述參數值,回收率均無明顯增加。因此,實驗選擇最佳萃取溫度為60 ℃,萃取時間為30 min,萃取溶劑體積為30 mL。
2.5線性關系與檢出限
在優化的色譜-質譜條件下進行測定,以4種BUVSs在MRM模式下定量離子的峰面積(y)對相應的質量濃度(c,μg/L )進行線性回歸計算,得到線性方程和相關系數(r2)。分別以信噪比S/N=3和S/N=10時的質量濃度確定方法的檢出限(LOD)和定量下限(LOQ),結果見表3。4種BUVSs的相關系數均大于0.99,UV-350,UV-320,UV-328和UV-327的LOD分別為0.023,0.024,0.006 3,0.021 mg/kg,LOQ分別為0.078,0.081,0.021,0.072 mg/kg。
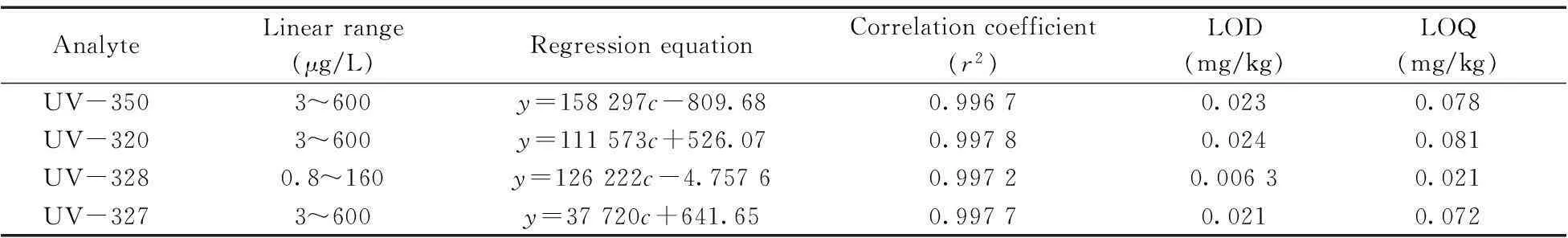
表3 UPLC-MS/MS方法的分析性能
2.6回收率與精密度
以陰性棉、錦綸為樣品,分別添加1倍LOQ、2倍LOQ、10倍LOQ 3個濃度水平的BUVSs,進行加標回收率和精密度實驗,每個濃度平行測定6次,結果如表4所示。在3個濃度水平下,不同基底紡織品中目標化合物的平均回收率為80.0%~102.9%,相對標準偏差(RSD)為1.2%~9.9%,方法的準確度和精密度能滿足紡織品中BUVSs的測定要求。
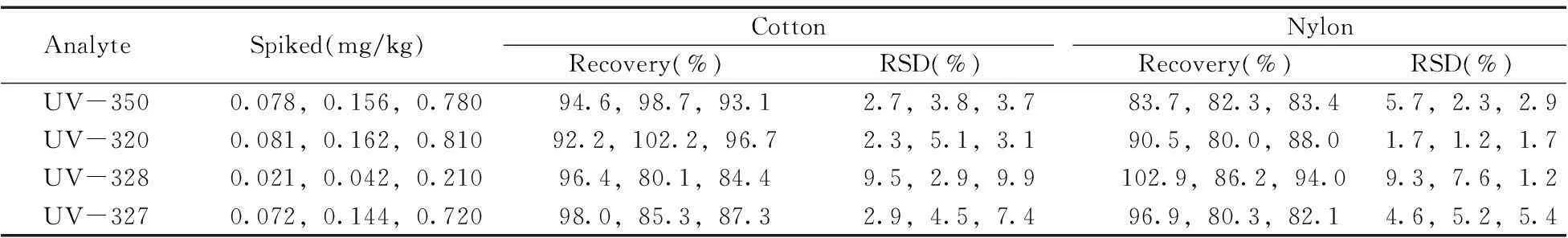
表4 方法的回收率及相對標準偏差(n=6)

圖2 樣品中UV-328的MRM圖Fig.2 UPLC-MS/MS chromatograms for UV-328 found in sample
2.7樣品分析
采用本方法對13個防紫外線紡織品樣品中4種BUVSs進行篩查測定,其中1個樣品定性檢出UV-328(見圖2),但其含量低于LOQ(0.021 mg/kg),其它樣品均未檢出4種BUVSs。這可能是因為樣品來源單一并集中在少數防紫外線紡織品樣品的緣故。
3結論
本文建立了超聲提取/超高效液相色譜-串聯質譜法測定紡織品中4種BUVSs的分析方法。該方法以正己烷飽和的乙腈為提取溶劑,超聲提取樣品中的待測組分,萃取液經過濾后進行超高效液相色譜-串聯質譜法多反應監測模式測定。方法簡便、快速、靈敏、準確,可用于紡織品中4種BUVSs的實際檢測,并可為制定相關檢測標準提供參考。
參考文獻:
[1]Nakata H,Shinohara R,Murata S,Watanabe M.J.Environ.Monit.,2010,12:2088-2092.
[2]Reiter S M,Buchberger W,Klampfl C W.Anal.Bioanal.Chem.,2011,400(8):2317-2322.
[3]Himmelsbach M,Buchberger W,Reingruber E.Polym.Degrad.Stab.,2009,94(8):1213-1219.
[4]Huang F Q,Li Q L,Yang D J,Feng X N,Zheng X.ChinaDyeing&Finishing(黃方千,李強林,楊東潔,馮西寧,鄭雄.印染),2011,37(20):47-52.
[5]Cancilla D A,Christopher Baird J,Geis S W,Corsi S R.Environ.Toxicol.Chem.,2003,22(1):134-140.
[6]Schlumpf M,Cotton B,Conscience M,Haller V,Steinmann B,Lichtensteiger W.Environ.HealthPerspect.,2001,109(3):239-244.
[7]Schlumpf M,Schmid P,Durrer S,Conscience M,Maerkel K,Henseler M,Gruetter M,Herzog I,Reolon S,Ceccatelli R,Faass O,Stutz E,Jarry H,Wuttke W,Lichtensteiger W.Toxicology,2004,205(1):113-122.
[8]Jarma H,Scrimshaw M D,Wmiams R J,Churchley J,Sumpter J P.Environ.Sci.Technol.,2011,45(9):3858-3864.[9]Jungclaus G A,Lopez-Avila V,Hites R A.Environ.Sci.Technol.,1978,12(1):88-96.
[10]Ruan T,Liu R Z,Fu Q,Wang T,Wang Y W,Song S J,Wang P,Teng M,Jiang G B.Environ.Sci.Technol.,2012,46(4):2071-2079.
[11]Carpinteiro I,Ramil M,Rodríguez I,Nogueira J M F.J.Sep.Sci.,2012,35(3):459-467.
[12]Lai H J,Ying G G,Ma Y B,Chen Z F,Chen F,Liu Y S.Environ.Sci.:Proc.Imp.,2014,16(3):558-566.
[13]Carpinteiro I,Abuín B,Rodríguez I,Ramil M,Cela R.J.Chromatogr.A,2010,1217(24):3729-3735.
[14]Carpinteiro I,Abuín B,Ramil M,Rodríguez I,Cela R.Anal.Bioanal.Chem.,2012,402(1):519-527.
[15]Kim J W,Isobe T,Ramaswamy B R,Chang K H,Amano A,Miller T M,Siringan F P,Tanabe S.Chemosphere,2011,85(5):751-758.
[16]Kim J W,Ramaswamy B R,Chang K H,Isobe T,Tanabe S.J.Chromatogr.A,2011,1218(22):3511-3520.
[17]Kim J W,Isobe T,Malarvannan G,Sudaryanto A,Chang K H,Prudente M,Tanabe S.Sci.TotalEnviron.,2012,424(4):174-181.
[18]Liu R Z,Ruan T,Wang T,Song S J,Guo F,Jiang G B.Talanta,2014,120(1):158-166.
[19]Wang J C,Zhang H J,Chen J P,Zhang L.Chin.J.Chromatogr.(王金成,張海軍,陳吉平,張玲.色譜),2014,32(9):913-918.
Simultaneous Detection of Four Benzotriazole UV Stabilizers in Textiles by Ultra-high Performance Liquid Chromatography-Tandem Mass Spectrometry
ZHU Feng1,2*,WEI Min1,2,SHI Dian-wang1,2,XUE Jian-ping1,2
(1.Fujian Provincial Fiber Inspection Bureau,Fuzhou350026,China;2.Fujian Provincial Key Laboratory of Textiles Inspection Technology,Fuzhou350026,China)
Abstract:A method was developed for the simultaneous determination of four benzotriazole UV stabilizers(BUVSs) including UV-350,UV-320,UV-328 and UV-327 in textiles by ultra-high performance liquid chromatography-tandem mass spectrometry(UPLC-MS/MS) combined with ultrasonic extraction(USE).The samples were extracted with acetonitrile saturated with n-hexane under ultrasonic assistance.The separation was performed on an Eclipse Plus C18column(2.1 mm×50 mm,1.8 μm) using 0.01 mol/L ammonium formate-methanol as mobile phase.Identification and quantification were achieved by UPLC-MS/MS coupled with electrospray ionization(ESI) source in positive ion mode and multiple reactions monitoring(MRM) mode,and the concentration of each analyte was calibrated by the external standard method.The result indicated that the calibration curves of four BUVSs showed good linear relationships between peak areas and certain concentration ranges with correlation coefficients(r2) greater than 0.99.The limits of detection for UV-350,UV-320,UV-328,UV-327 were 0.023,0.024,0.006 3,0.021 mg/kg,respectively.The limits of quantitation were 0.078,0.081,0.021,0.072 mg/kg,respectively.The recoveries at three spiked concentration levels were in the range of 80.0%-102.9% with relative standard deviations(RSDs) of 1.2%-9.9%.The developed method was successfully applied in the determination of BUVSs in textiles.
Key words:ultra-high performance liquid chromatography-tandem mass spectrometry(UPLC-MS/MS);benzotriazole UV stabilizers(BUVSs);textiles;ultrasonic extraction(USE)
收稿日期:2015-09-29;修回日期:2015-11-06
基金項目:福建省質量技術監督局科技項目(FJQI2014020)
*通訊作者:朱峰,碩士,工程師,研究方向:有毒有害物質色譜質譜分析及其質量控制,Tel:0591-83595610,E-mail:zhufengdr@163.com
doi:10.3969/j.issn.1004-4957.2016.04.007
中圖分類號:O657.63;TQ047.4
文獻標識碼:A
文章編號:1004-4957(2016)04-0414-06
猜你喜歡
紡織標準與質量(2022年1期)2022-07-12 06:01:02
化工管理(2021年7期)2021-05-13 00:45:12
知識經濟·中國直銷(2018年4期)2018-04-18 12:04:49
紡織科學研究(2017年8期)2017-09-05 09:46:35
產業用紡織品(2016年9期)2016-07-07 15:19:44
紡織服裝流行趨勢展望(2016年4期)2016-05-04 03:51:13
紡織科技進展(2015年1期)2015-11-28 05:56:12
中國洗滌用品工業(2015年9期)2015-02-28 19:03:06
現代紡織技術(2015年6期)2015-02-28 14:03:23
流行色(2005年4期)2005-04-29 00:44:03
