第五章 三大合成催化
2016-05-17 09:11:01孫文華
工業催化 2016年2期
關鍵詞:催化劑
孫文華
?
第五章三大合成催化
孫文華
(中國科學院化學研究所,北京 100190)
5.1引言
5.1.1合成高分子的歷史發展
伴隨人類出現,就開始了使用天然高分子材料的歷史,包括使用樹木和植物搭建窩棚及幫助攀爬等,在進化過程中開始使用藤蔓植物和蘆葦、樹皮和棉花等的編織物,直到19世紀中葉,開始跨入天然聚合物的化學改性時代。1839年Charles Goodyear使用硫化反應使天然橡膠更具彈性和實用性[1];1865年John Wesley Hyatt使用樟腦增塑硝化纖維素[2],并于1870年實現了硝化纖維塑料的工業化。20世紀進入合成高分子時代,1907年Leo Henricus Arthur Baekeland首次實現了熱固性酚醛樹脂的合成[3],成為20世紀20年代實現工業化的第一個合成塑料。開啟高分子科學研究,公認是Hermann Standinger教授1920年定義高分子是由結構單元重復并通過普通共價鍵彼此連接而形成的長鏈分子,奠定了高分子科學發展的基礎。合成高分子先驅Wallace Hume Carothers 1930年4月與合作者Arnold M Collins使用分離的氯丁二烯做單體合成了氯丁橡膠,被認為是第一種合成橡膠;不僅如此,1934年Wallace Hume Carothers集中開展纖維的合成研究,利用二胺和二酸進行縮合反應制備了聚酰胺,基于此,與Gerard Berchet合作于1935年2月使用1,6-己二胺與己二酸反應制備了6,6-聚酰胺(頗具政治色彩的是由于6,6-聚酰胺能夠制備高彈纖維,展示了蠶絲的功能,在20世紀30年代,日本是蠶絲制品最大生產國。Wallace Hume Carothers所在的DuPont公司期望6,6-聚酰胺的纖維能夠實現對于日本蠶絲的替代,采用“Now You Lose Old Nippon”的縮寫Nylon命名了6,6-聚酰胺)。Wallace Hume Carothers的研究充分展示了合成高分子的兩類重要反應:加成反應與縮合反應。與此同時,由于德國Standinger H教授(1932年)大分子長鏈結構理論的確立和前蘇聯謝苗諾夫H H的鏈式聚合理論(1934年)的提出,為加成高分子合成奠定了科學基礎。英國帝國化學工業公司的Fawcett E W在英國皇家化學會Faraday會議上報告了乙烯高壓聚合制備聚乙烯[4],該技術于1939年實現了百噸產業化生產;當時擁有相關技術的還有DuPont和UCC公司,聚乙烯材料生產和應用還僅僅作為軍用物資,僅在第二次世界大戰后獲得更多公司推廣。量變的過程必然帶來質的跨越,20世紀50年代初,德國科學家Karl Ziegler發展了鈦配位乙烯(及烯烴)聚合催化劑,意大利科學家Giulio Natta快速推進了鈦配位丙烯聚合及產業化。
20世紀50年代末開始的大規模聚烯烴產業化,使得價廉質優的高分子材料更具民用價值和意義,更為廣泛的高聚物研究與應用成為當時最具發展潛力的學科之一。目前合成高分子的塑料、合成橡膠和合成纖維使用總量超過3億噸,成為提高人類生活水平與減緩地球資源消耗的重要保障材料,成為人們衣、食、住、行和工農業生產必要的物資。
5.1.2合成高分子材料的重要價值
工農業生產與現代服務業發展的目標是提高人類生活水平;不僅如此,為了保障后代的繁衍生息,需要減緩天然資源的使用與保護環境,求得持續與長久發展。盡管民眾錯覺認為“合成高分子材料是環境污染的罪魁禍首”,設想一下沒有合成高分子材料所提供的支持和保障,維持目前人類三分之一人口生活水平的天然物質基礎會顯得異常困難,地球資源的消耗和溫室效應都會呈級數演變。
伴隨人類和動物從穴居到窩棚和住宅的進化,快速地消耗著石材和樹木,造成森林快速減少,甚至成片消失;空氣中溫室氣體濃度加大,加速了冰川消融和海平面的升高,慢慢吞噬著人類長期居住的家園。這類環境的破環,才真正無法再生。與之對應,高分子材料被冠以“環境的公敵”,卻在當今建筑材料中發揮著越來越重要的作用,提供了輕量化、高強度、隔熱、抗沖擊和抗震的優良材料,已經成為臨時建筑的框架與墻體和固定建筑中防滲漏、涂料、密封劑、黏合劑、給排水、電訊、隔音與保暖等不可缺少的材料,更是快速發展的城市中高層建筑必須的輕量建材。科學總是“雙刃劍”,高分子建材和家裝材料存在易燃與老化及事故中有毒煙霧的缺點備受批評,其替代鋼鐵、水泥與木材的功能,直接保護了自然資源和提高了人類的居住環境與生活品質。材料科學家仍然在使用摻雜和共混持續地提高高分子材料的性能和克服其存在的缺點。
比居住條件更為重要的是“衣”,人類通過天然纖維的應用提高了審美和生活的品質,這些天然纖維包括植物纖維的棉與麻以及動物纖維的毛與蠶絲等。在地球人口數量超過60億的當下,天然纖維已經遠遠無法滿足量的供給,更無法滿足人們對于衣物的色澤、保暖、堅固耐磨、防霉防蛀和免洗免燙等特點的追求;即使熱戀天然織物的人,也不難發現自己的“棉、麻、毛和蠶絲”衣物很難占到兩成,甚至很有可能標記為“天然纖維”的衣服中仍然含有合成高分子材料。不僅如此,高分子纖維鮮艷的光澤和美感備受時尚界的青睞和更具商業價值;例如:尼龍長襪具有棉質感和更具保暖與易洗滌的腈綸休閑服裝,透氣性能與干爽效果更佳的維尼綸內衣織品等,無疑撼動了天然高分子纖維的特征價值。除高分子纖維之外,裝點普通百姓衣物的還需要飾品與紐扣,其基本原料更多出自高分子合成材料。市面上流行的仿珍珠紐扣與飾品取材于聚酯,仿玉制品基于聚酯或者聚甲醛樹脂,這些飾品有更光滑的表面和靚麗的色澤,甚至可以比較便利地包裹小昆蟲與植物做成琥珀飾品,給人更高檔的色感和沖擊力。值得補充說明,高分子纖維的衣物和飾品使用更具耐久性,沒有高分子纖維對于衣物和飾品的基石,天然材料無法滿足現在人類對于衣物的欲望需求的十分之一。
與人類生活品質密切相關的還有“行”,高分子材料在交通工具部件和資源節約中的價值更是毋庸置疑。包括汽車車體與內外加護和裝飾材料,飛機、輪船和火車的框架與部件以及內外裝飾材料。對于鋼鐵與木材的替代,以及高分子材料輕量化帶來的動力消耗大幅的減少,都對自然的保護起了巨大作用!盡管在部分產品中暴露了強度不足的安全性和燃燒性問題,碳纖維材料無論在汽車和飛機甚至空間衛星制造中都成為強度高和耐久與耐熱性能俱佳的材料。高分子材料使用的范圍與性能要求可以滿足制造不同層次交通工具的需求,人們不能都采取最高材料標準的關鍵還是材料制備的工藝與技術過程的能耗問題。無疑,使用更多高分子材料制造交通工具成為材料科學與制造業發展的主流。
不僅如此,如果說“高分子材料甚至提高了你的飲食質量”,你會如何反應?高分子材料并非用于食用,但是在食品儲運與保鮮中高分子材料的作用至關重要,隔斷了細菌傳播和滋生的條件,甚至極大地減少了交叉感染的可能,保護了身體,提高了生活質量,滿足了人類需求。大家在爭論一次性餐具和哪種做法更保護自然環境。拿一個常被詬病的“一次性塑料杯和陶瓷杯”招待客人哪個更環保進行比較,即使不計算清洗和用水,制備一個陶瓷杯的能耗是一次性塑料杯能耗的兩千倍!至于其環境的負面影響,這些高分子包裝和容器完成功能后最有效的利用手段就是焚燒和作為能源的原料;此外,也有再降解成為有機單體和產業化工程裝置的研究,顯得更具吸引力與持續發展前景。
5.1.3合成高分子材料分類
高分子材料種類眾多,學術界和產業界有很多不同的分類方式,尤以按照材料使用時的形態分類被廣泛接受,分為“塑料、纖維和橡膠”三大類。毋容置疑,涂料和膠粘劑作為合成高分子材料越來越受到重視,但是在催化科學為主的教材中,這里僅簡單介紹“塑料、纖維和橡膠”。
5.1.3.1塑料
塑料又稱為樹脂,“樹脂”顧名思義,是植物滲(泌)出物的無定形(半)固體有機物;天然樹脂的使用可以追溯到數千年前,最早使用的天然樹脂是松香、蟲膠和琥珀等。盡管現代塑料工業大家比較接受的是始于20世紀30年代,但可以追溯到19世紀中葉為了尋找天然樹脂的代用品,美國人海厄特J W在濕潤的硝酸纖維素中加入樟腦和少量酒精制成了一種可塑性物質,能夠熱壓下成型,命名為“賽璐珞”替代蟲膠,用于馬車和汽車的擋風玻璃和后來作為電影膠片的材料,并于1872年建廠生產賽璐珞;1903 年德國人艾興格林A發明了不易燃燒的醋酸纖維素和注射成型方法,1905 年德國拜耳股份公司進行工業生產。與此同時,美國的Albang Dental Plate公司于1870年最早研究酚醛樹脂的合成,后由美國Bakelite(酚醛樹脂)公司于1909年將該技術實現產業化生產。比美國更早進行第一個工業化酚醛樹脂生產的是1902年布盧默(Blumer L)用酒石酸催化酚和醛縮合制得命名為“Laccain”的酚醛樹脂,遺憾的是沒有形成規模;被稱為酚醛樹脂創始人的美國科學家巴克蘭(Baekeland)是1905年才開始用苯酚和甲醛來合成樹脂的研究,并于1909年首次提出酚醛樹脂“加壓和加熱”的熱固性塑料,奠定了其科學威望,并在1924年擔任美國化學會主席。1910年在柏林呂格斯工廠建成了通用酚醛樹脂公司,更加提升了酚醛樹脂的批量生產;應該說在20世紀40年代前,酚醛塑料約占塑料產量的三分之二,是最主要的塑料品種,廣泛用于電器、儀表、機械和汽車工業。
在跨入現代塑料工業過程中,還值得提及1930年德國法本公司進行工業聚苯乙烯生產,1931年美國羅姆-哈斯公司以本體法生產聚甲基丙烯酸甲酯(有機玻璃);以及20世紀40年代用乳液法生產聚氯乙烯。然而,具有現代塑料工業里程碑的研究是1933年英國卜內門化學工業公司(ICI)兩位研究人員Reginald Gibson和Eric Fawcett在進行乙烯與苯甲醛170 ℃加壓反應時,發現聚合釜壁上有蠟質固體存在,發明了聚乙烯,并于1939年實現高壓氣相本體法生產低密度聚乙烯;該技術在第二次世界大戰中由多國公司生產聚乙烯,當時聚乙烯還僅僅限于戰備物資使用。塑料獲得普及和大量應用還是在1953年聯邦德國Karl Ziegler(齊格勒)教授領導的實驗室發明烷基鋁活化四氯化鈦常壓催化乙烯聚合,并在1955年聯邦德國赫斯特公司首先采用低壓下制備高密度聚乙烯;受齊格勒研究的啟發,意大利塑料改性共混研究的學者納塔G抓住機會突擊丙烯聚合研究,其技術于1957年由意大利蒙特卡蒂尼公司實現了聚丙烯工業生產。此后的發展則一發而不可收,塑料世界總產量從1904年的10 kt,到1956年達到3.4 Mt,2012年使用量超過了220 Mt(見圖5-1)。大品種塑料中包括聚烯烴、聚苯乙烯、聚碳酸酯、ABS樹酯、聚苯醚和聚酰亞胺等;其中聚烯烴占了塑料總量的近三分之二。
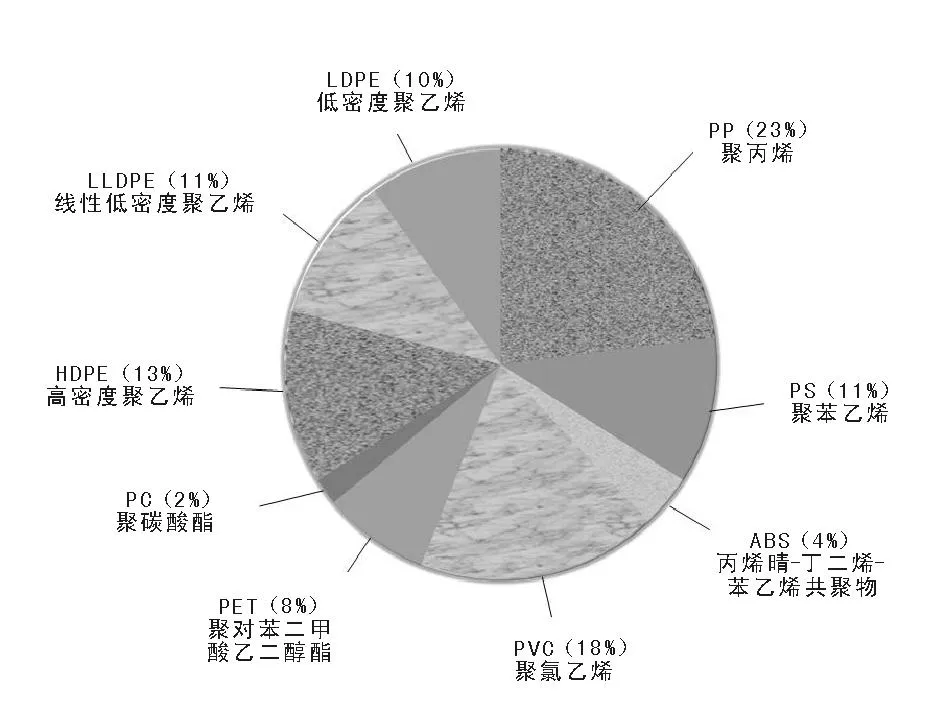
圖 5-1 全球塑料產品消費量(2012年超過220 Mt)
5.1.3.2合成橡膠
1860年,威廉斯C G在熱裂解天然橡膠產物時收集到了異戊二烯,并且發現異戊二烯在空氣中放置會變成白色彈性體,布查德 G又在1879年重復了該技能。1900年孔達科夫И Л采用2,3-二甲基-1,3-丁二烯聚合制備彈性體,2,3-二甲基-1,3-丁二烯在70 ℃熱聚合歷經 5個月后獲得彈性體稱為甲基橡膠W,而在(30~35) ℃聚合歷經3~4個月后制成略硬的橡膠稱為甲基橡膠H。在第一次世界大戰期間,德國的海上運輸被封鎖,切斷了天然橡膠的輸入,該技術于1917年在德國獲得工業化生產;然而,其性能遠比天然橡膠差得多,停戰后隨即停產,僅生產了2.3 kt。前蘇聯列別捷夫 C B利用酒精轉化成的丁二烯采用鈉催化液相本體聚合,制得了丁鈉橡膠,在1931年建成萬噸級生產裝置;同一時期,德國人從乙炔出發合成丁二烯,也實現了丁鈉橡膠的制備,德國法本公司1935年首先實現丁腈橡膠生產和1937年在布納化工廠建成丁苯橡膠工業生產裝置,而丁腈橡膠是一種耐油橡膠,目前仍是作為特種橡膠使用。第二次世界大戰的急需和當時日本占領了天然橡膠主產地馬來西亞,促進了橡膠產業的快速發展,世界合成橡膠產量從 1939年的23 kt劇增到1944年的885 kt。不僅如此,還出現了許多特種橡膠新品種,包括美國通用電氣公司1944年生產的硅橡膠,同期德國和英國分別生產了聚氨酯橡膠。
隨著齊格勒-納塔烯烴聚合工業化,石油化工裂解制備乙烯與丙烯過程中產生了大量二烯單體,配合新型催化劑研發的溶液聚合技術更有效地控制橡膠分子的立構規整度,提高了橡膠性能,使合成橡膠工業進入一個嶄新的階段。主要品種包括:接近天然橡膠性能被稱為合成天然橡膠的高順式1,4-聚異戊二烯(簡稱異戊橡膠),高反式1,4-聚異戊二烯(簡稱合成杜仲膠),順式1,4-聚丁二烯橡膠(簡稱順丁橡膠)以及丁腈橡膠、氯丁橡膠和丁苯橡膠等;此外還有乙烯與丙烯共聚制備的乙丙橡膠和聚氨酯彈性體等。到20世紀70年代后期,合成橡膠已基本可以替代天然橡膠制造各種輪胎和制品,而且特種合成橡膠具有天然橡膠無法達到的性能與品質。目前合成橡膠的年產量約7 Mt。
5.1.3.3合成纖維
在合成高分子中另一個重要分支就是“合成纖維”。1913 年,德國人Klatte F合成了聚氯乙烯纖維,該技術于1934年由德國IG化學公司實現工業化生產,是最早的合成纖維,然而,由于耐熱性差阻礙了其應用與發展。同期美國科學家、縮聚合成高分子的奠基人Carothers W H博士于1935年使用己二胺和己二酸縮聚合成了聚酰胺66,DuPont 公司在1938年實現了中試生產,1939年紡絲成功聚酰胺66纖維,俗稱尼龍;當1940年投放市場,立刻成為世界上爭相搶購的織物,成為第一種大規模生產合成纖維。1941年英國“Calico Printers Association”公司的Whinfield J R和Dickson J T成功地使用對苯二甲酸與乙二醇實現縮聚,并于1944年成功實現熔體紡絲;該技術于1947 年由英國ICI實現產業化生產聚對苯二甲酸乙二醇酯纖維,在1953年DuPont 公司從英國購買了專利進行聚酯纖維“Dacron”的大規模生產。聚酯纖維成本低和用途廣泛,至1972 年聚酯纖維產量超過聚酰胺,成為最大量和應用最廣的合成纖維。
然而,合成纖維另一個平行的增長在于Ziegler-Natta催化劑制得廉價的聚乙烯和聚丙烯成為新的紡絲材料,意大利Montefibre公司自1960年就實現了聚丙烯纖維的工業化,吸引了國際產業界的競爭。2013年合成纖維84.5 kt,其中聚烯烴纖維占52.7 kt,其他重要合成纖維還有聚酯、聚酰胺、聚丙烯腈等,合成纖維的快速發展也擠壓了棉花的生產與銷售空間,使天然纖維呈現萎縮局面。
此外,作為一般信息,還需特別提及兩種特種纖維:碳纖維和超高分子量聚乙烯纖維。碳纖維質量比鋁材輕,強度卻高于鋼鐵,并且在有機溶劑、酸、堿中不溶不脹及耐腐蝕和模量高等特點,成為航空和國防軍工的重要物資,并在民用奢侈品和耐用品市場開始使用,呈現出巨大潛力。碳纖維生產主要分為聚丙烯腈基碳纖維、瀝青基碳纖維等,其生產具有戰略意義,備受航空與軍工企業重視;國家多次組織重點攻關和產學研聯合研究,實現了丙烯腈聚合和紡紗制備原絲,在將原絲放入氧化爐中300 ℃進行氧化后,再升溫到(1 000~2 000) ℃進行碳化工序處理,制備碳纖維,在國內已有吉化公司實現了規模化和批量生產(其他民營公司也采用相似的技術工藝)。超高分子量聚乙烯纖維(Ultra High Molecular Weight Polyethylene Fiber,簡稱UHMWPE),又稱高強高模聚乙烯纖維,是目前世界上比強度和比模量最高的纖維,其分子量在1×106~5×106的聚乙烯凍膠紡絲制成的纖維,具有抗紫外線輻射,防中子和γ射線,比能量吸收高、介電常數低、電磁波透射率高等特點;最初集中用于防彈衣、防彈頭盔、軍用設施和設備的防彈裝甲、航空航天等軍事領域,近年來用于纜繩、防切割手套和漁網(養魚箱)等制備。其技術核心是乙烯聚合中對于超高分子量聚乙烯分子量和分子量分布的有效控制,以及凍膠(干法和濕法)紡絲品質控制。我國在2008年奧運前作為重點攻關項目由原北京東方化工廠助劑二廠(現并入中國石化燕山分公司)聯合中國科學院化學所使用中國石化奧達催化劑公司催化劑實現了萬噸級產業化,國內多家公司成功用于紡絲和制備相關產品,其防彈衣、防彈頭盔與防彈裝甲保障國家需求,還出口西亞諸國。
5.2高分子材料合成中典型催化問題
無論現代化工還是高分子材料的合成過程無疑需要催化工藝,降低日益增加的勞動力和場地成本,而且在提高生產效益的同時,改善了產物的品質。在高分子材料合成按照反應類型分為加成聚合和縮聚反應,需要給出必要的實例進行探討。在上面介紹中不難看出,合成高分子材料基于其功能分為塑料、合成纖維和橡膠。然而,基于科學本質,科學研究關心原料和反應過程以及所得材料的微結構,產物材料的微結構控制了其宏觀性能和應用領域與范圍。
目前,占據塑料產量市場三分之二和合成纖維中超過六成的都是基于聚烯烴材料,而且聚烯烴微結構調控延伸到橡膠替代的彈性體材料。聚烯烴材料以其“成本低、重量輕和易加工”的特性,而且僅僅限于碳氫化合物的組成保證其使用后仍可作為燃料的環保品質,成為重點推廣的材料;不僅如此,聚烯烴樹脂生產能力與技術水平也是衡量一個國家石化產業發展水平的重要標志。聚酯其組成限于碳氫氧,其制品在完成使用功能后無論降解還是焚燒的產物都具有環境耐受力,而且廣泛用于塑料制品與合成纖維的原材料。橡膠作為不飽和鍵存在、碳氫組成的高分子材料,具有無法替代的性能和應用。聚烯烴、聚酯和橡膠材料使用量占到目前合成高分子材料的九成,其催化技術無疑主導了高分子材料合成的效益和未來發展趨勢,掌握了這三類催化技術,不難展開進行其他高分子材料的催化研究。為此,我們在重點介紹聚烯烴催化的基礎上,對聚酯和橡膠催化進行必要闡述。
5.2.1聚烯烴材料及其催化劑
自1939年聚乙烯開始工業化生產以來,聚烯烴的發展至今已有70多年的歷史,最初的烯烴采用醇脫水制備;20世紀50年代,石油裂解制備烯烴技術極大地提升了石油化工產品的附加值,更重要的是改善了人們的日常生活,被認為是20世紀最偉大的發現之一。聚烯烴具有優良的材料性能,使用中無毒和穩定性能高;通過共聚與改性可制備特種專用樹脂,具有高抗沖、高耐熱、高透明、低熱封溫度、導熱、導磁或高屏蔽等性能。雖然聚苯乙烯科學上分類為聚烯烴,但是通常俗稱的聚烯烴還是專指聚乙烯和聚丙烯及其共聚物;在石油裂解制備烯烴單體時,總是乙烯產量高于丙烯,因而從20世紀中期聚丙烯為主發展到聚乙烯更受到關注。作為聚烯烴性能與分類的知識,其本質還是微結構“支化度多寡與長短,分子量高低與分布寬窄”影響到宏觀指標“密度和熔融指數”與材料性能。以乳白色半透明蠟狀固體的聚乙烯為例,分為高密度、中密度、低密度和超低密度聚乙烯。密度在(0.941~0.965) g·cm-3的高密度聚乙烯(HDPE)通常由Ziegler-Natta催化劑或鉻系(Phillips)催化劑制備,支鏈化程度最低,結晶度較高,強度和抗老化性能優于聚丙烯,使用溫度優于聚氯乙烯,廣泛用于注塑、吹塑、擠塑和旋轉成型制品加工;此外,超高分子量聚乙烯(UHMWPE)也是高密度聚乙烯中的一員,其密度甚至接近1.0 g·cm-3,采用Ziegler-Natta催化劑和低溫(60 ℃)聚合制備,分子量超過100萬,用于制備高強和高模聚乙烯纖維,也用于高強板材制備。密度在(0.916~0.940) g·cm-3的中密度聚乙烯(MDPE),結晶度約75%,拉伸強度較高密度聚乙烯差,其剛性、耐磨性和透氣性介于高密度聚乙烯和低密度聚乙烯之間,適于用作擠塑管材、蒸煮袋的內襯薄膜和包裝材料等。密度在(0.910~0.925) g·cm-3的低密度聚乙烯(LDPE),通常采用高溫和高壓自由基聚合制備,常稱為高壓聚乙烯,分子量一般在50 000~500 000,結晶度較低,具有良好的耐低溫性能,可用于(-60~-80) ℃的工作溫度,電絕緣性能好,是擠塑管材和電線、電纜的重要原料。密度更低的材料稱為線型低密度聚乙烯(LLDPE)以及超低密度聚乙烯(VLDPE),通常由茂金屬(metallocene)或Ziegler-Natta催化劑進行乙烯與α-烯烴共聚合制備,由于α-烯烴形成側鏈造成結晶度低和密度小,但具有良好的表面光澤性和低溫韌性高的特點,其高模量、抗彎曲和耐應力開裂性的優點廣泛用于吹膜和包裝材料。
我國第一套聚烯烴生產裝置是20世紀70年代引進日本三井的聚丙烯成套裝置在燕山投產;聚烯烴工業發展模式一直是關鍵技術依賴進口,因而,買方市場的后果是大量裝置滿足的僅僅是中低檔聚烯烴材料的需求。考慮到國際聚烯烴企業對于我國企業的聯合抵制,許多引進聚合技術和裝置在引進之初并非先進,造成在我國成為聚烯烴材料生產大國和Ziegler-Natta催化劑重要生產國的當下,消耗量超過四分之一的高性能聚烯烴樹脂仍然依賴進口。關鍵根源在于基礎研究發展的薄弱和技術開發的滯后,導致我國缺少自主知識產權的“聚烯烴催化劑和聚合工藝”,因而,迫切需要催化劑和催化工藝上創新研究和技術突破。
5.2.1.1鉻系(Phillips)催化劑
負載鉻系催化劑是基于硅膠或者二氧化硅與氧化鋁復合顆粒作載體負載鉻,由于是Phillips公司設在俄克拉荷馬州的研究室成員J Paul Hogan和Robert L Banks于1951年的發明[5],并且由Phillips公司于1955年實現連續法工業生產,又稱為Phillips催化劑。盡管出于環境保護的需求,國際上對于廢除鉻使用的呼聲很高,該催化劑體系仍然生產國際大量消耗的高密度聚乙烯份額中的四成,其原因在于鉻系催化劑所得高密度聚乙烯具有較寬的分子量分布,在滿足材料性能要求的同時,具有良好的加工性能,備受成品制造者的青睞。
制備鉻系催化劑,通常采用硅膠顆粒懸浮在溶劑中,加入水溶良好的鉻酸銨,獲得硅球吸附三氧化二鉻的顆粒(圖5-2)[6],然后高溫焙燒制得具有催化聚合活性的鉻系催化劑,其中鉻在負載催化劑中的負載質量分數為0.2%~2%。

圖 5-2 氧化鉻在硅膠載體表面的負載
20世紀70年代,六價鉻被確認為致癌物(因而,國內尚存部分實驗室使用重鉻酸鉀洗液用于清洗玻璃儀器的做法值得停止),為了減少鉻系催化劑制備中操作者與六價鉻的接觸,紛紛采用三價鉻鹽替代[7],特別是產業界最常用的醋酸鉻,在負載催化劑焙燒過程中很容易將三價鉻氧化為六價鉻,其陰離子則很容易揮發掉。大量的研究證實,鉻離子變價轉化比較容易,考慮到乙烯聚合是一個相對強的還原氣氛,科學家使用光譜跟蹤驗證了分別使用一氧化碳和氧氣環境中鉻的氧化與還原狀態,認為獲得了六價鉻和二價鉻的證據(圖5-3[6]),其他價態組分中間體存在值得推測。

圖 5-3 CO/O2氣氛下六價鉻與二價鉻間的還原與氧化
盡管各種價態的鉻都被證實或者準確地講是猜測為催化乙烯聚合的活性組分,也被用作解釋鉻系催化劑所得聚乙烯分子量分布較寬的根源。六價鉻是桔紅色,三價鉻是藍色,實驗表明,三氧化鉻(六價鉻)在超過200 ℃處理時很容易轉化為低價鉻[8];而實際操作中,工業鉻系催化劑使用前都是需要焙燒處理才能充分發揮其催化聚合活性,因而,近些年的研究逐漸接受和確認三價鉻為催化聚合的活性中心。最新的研究更加驗證了20世紀60年代Cossee提出的催化聚合機理[9]。

圖 5-4 乙烯聚合鏈增長和終止的Cossee機理
該機理認為,乙烯作為還原氣氛下,高價鉻首先還原成為低價鉻,配位吸附的乙烯轉化為烷基或者氫在鉻上鍵聯,形成催化中間體。基于這個烷基鍵聯在鉻上的催化中間體(圖5-4),單體乙烯進行配位,發生持續乙烯插入反應形成了長鏈聚乙烯鏈;一旦這個聚乙烯鏈足夠長,新的配位乙烯單體與之發生氫遷移,就產生了最終的聚乙烯以及新的烷基聯接在鉻上的新催化中間體;新的催化中間體可以持續誘導乙烯進行新的聚合反應。盡管也有很多文獻報道了 “鉻鍵聯烷基”和“鉻鍵聯氫”催化中間體的證據,但科技界普遍接受這種結論。這類催化中間體如何形成成為普遍關注的科學問題,畢竟鉻系催化劑可以不采用烷基金屬助催化劑就可以直接實現乙烯聚合。
一種解釋是硅膠中羥基的氫轉移到鉻上,形成“鉻鍵聯氫”的催化中間體,然后配位乙烯和插入反應形成“乙基鉻”催化物種,持續進行乙烯單體配位和插入獲得聚乙烯(圖5-5)[10]。
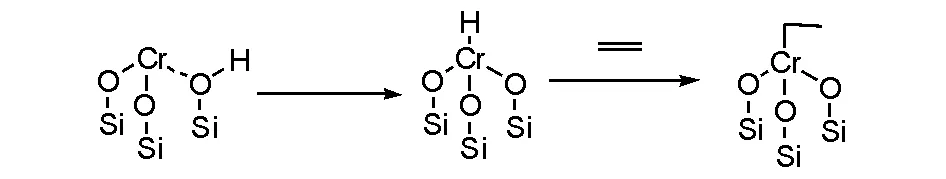
圖 5-5 基于硅膠羥基氫引發的鏈增長
另一種機理解釋(圖5-6)是基于二價鉻活性物種配位乙烯形成的乙叉基鉻(乙基卡賓鉻化合物),再與乙烯配位插入形成鉻雜環丁烷,再進行1,3氫遷移形成烷叉基鉻,重復乙烯配位插入,在進行2,3氫遷移時給出含有雙鍵的聚乙烯(或齊聚物)和二價鉻活性物種[11]。
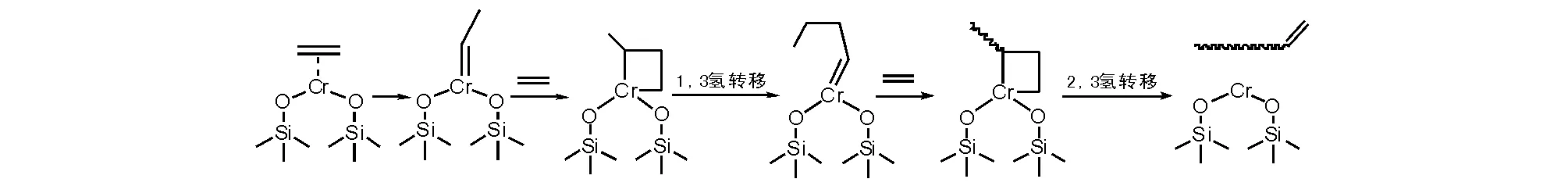
圖 5-6 基于二價鉻烷叉基乙烯插入成環鏈增長機理
還有一種機理解釋也需要特別關注,就是認為催化過程中形成了鉻雜戊烷中間體活性中心(圖5-7)[12]。成環機理中給出幾個重要信息能夠幫助解釋鉻系催化劑催化乙烯三聚制備己烯:氫遷移形成端烯基中間體以及丙烯基配位的催化中間體,而丙烯基中間體可以轉化形成雙鍵位移到內部的烷烴,這也是為什么所得聚乙烯未必能夠清晰測試其雙鍵的位置。
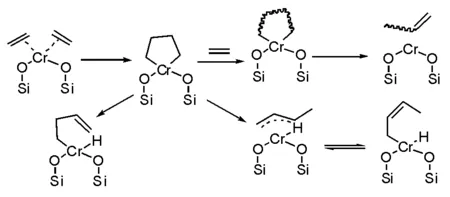
圖 5-7 基于鉻雜環烷基與丙酰基鉻催化中間體的機理
基于鉻系催化劑實用價值和巨大的商業利益,產業界仍會持續進行改良研究。由于鉻潛在致癌的問題,催化劑在聚烯烴中的殘留必將影響到該類聚乙烯樹脂的使用范圍在其被替代前,提高鉻系催化劑效率和性能將是永恒的研究主題。同時,仍有值得科學界去解決的謎團,這類基礎研究顯得更艱巨,無論反應機理和催化中間體的研究,目前計算化學的研究顯示了新的途徑,能否與實踐結果相符以及在什么程度上吻合仍然有待觀察。
5.2.1.2鈦系(Ziegler-Natta)催化劑
Ziegler-Natta催化劑是以德國科學家Ziegler K和意大利科學家Natta G兩人名字命名的最重要石化工業催化劑;自20世紀50年代問世以來,經過不斷研究和升級換代,催化性能不斷提高,推動了聚烯烴工業的迅猛發展,其生產規模不斷擴大,高性能聚烯烴樹脂層出不窮。然而,德國人教材中總是將這類催化劑稱為Ziegler催化劑;一個金屬有機背景的化學家Ziegler和頗具材料性能研究造詣的科學家Giulio Natta卻“涇渭分明”地研究這類催化劑對于乙烯和丙烯的聚合,正是兩人的重要貢獻分享了1963年的諾貝爾化學獎[在Ziegler研究組實現鈦催化乙烯聚合之后,同期開始了丙烯催化的研究,遺憾的是Ziegler研究組沒能得出丙烯聚合的結果。研究材料性能的Natta教授應邀參加了Ziegler組的催化乙烯聚合研究突破的發布會,會上提問中丙烯聚合研究的進展情況被告知“該催化劑(絕對)不能催化丙烯聚合”的結論觸動了Natta的研究興趣。也許就是“當事者迷,旁觀者清”,Natta回到意大利就物色合成和催化的合作者開展研究,在證實Ziegler的乙烯聚合研究結果之后,對于丙烯催化卻觀察到了蠟狀產物的少量產生——丙烯聚合實現了!帶著這個結果,Natta又去拜訪Ziegler和他的同事,側面問他們是否再試了丙烯催化;德國人告訴他,不間斷地試探并沒有獲得突破。Natta獲得堅強的信心和動力,決定盡快搞清兩個催化體系中的不同;但是,Natta仍然沒有告訴德國同事意大利學者實現了丙烯聚合。德國人良好的金屬有機合成與操作基礎保證了“Et3Al-TiCl4”體系純凈,而意大利學者使用反應體系密閉差,造成體系變化最終證實“Et2AlCl-TiCl3”催化丙烯聚合。科學的突破成就了兩位科學巨人,然而,也造成兩位巨人的個人恩怨,甚至1963年的諾貝爾化學獎頒獎時兩位都無法同臺,意大利學者在頒獎活動結束后才去領獎。這個故事也提供了創新的例證:新研究人員加入所觀察到的離奇研究結果或現象,不應該隨便忽視,更應該認真找到原因,也許正孕育一個偉大的發現]。
鈦系催化劑的研究起源于1953年德國化學家Ziegler K用Et3Al-TiCl4實現了乙烯的常溫、常壓聚合,得到了線性的高結晶度聚乙烯,即高密度聚乙烯[13-14]。1954年,意大利化學家Natta G[13]用Et2AlCl-TiCl3催化丙烯聚合,首次得到了全同立構的聚丙烯[15]。理性和客觀地講,Ziegler和Natta兩個課題組的工作是相互補充和競爭中獲得發展,缺少任何一部分都可能使Ziegler-Natta在聚烯烴工業發展的里程碑推遲。當時Ziegler研究組發現的催化體系,1 g鈦只能產生(1.5~3.0) kg聚乙烯,意味著聚乙烯中鈦殘留300×10-6~1 700 ×10-6;不僅所得聚合物有色,而且影響聚乙烯的穩定性,與使用的鉻系催化劑相比活性上沒有任何競爭力,而且當時還沒有人考慮鉻的毒性問題。與之對比,Natta研究組的催化體系受到更多關注,實現了丙烯聚合,由于是獲得全同立構的聚丙烯,就具有了快速結晶效果,結晶聚合物體現出更高的強度,當時鉻系催化劑無法實現。不僅如此,當時的烯烴(乙烯和丙烯)是通過醇脫水實現,丙烯制備更簡單和易于產業化,成為產業界競相研究和產業化的新材料。
科學研究中相同概念和主題研究間的啟發非常關鍵,鈦系催化劑很快借鑒了鉻系催化劑負載的概念,使用不同載體[MgCO3,SiO2/Al2O3(當時鉻系常用載體),其他金屬氯化物和氧化物等]進行氯化鈦負載制成的催化劑[16],活性獲得極大提高,發展了被稱為第二代鈦系催化劑(第一代鈦催化劑并沒有獲得應用),進入鈦催化乙烯聚合工業化生產階段。
對于負載鈦催化劑催化乙烯聚合的機理,一個趨向是接受Cossee機理(圖5-4),顯然可以接受。還有一個后來普遍接受被稱為Green-Rooney的機理[17-18],該機理與鉻系成環催化中間體(圖5-6)的機理類似;認為金屬活性中心周圍有充分的空位進行配位和形成金屬氫化物,成環與鏈增長獲得聚乙烯(圖5-8)。

圖 5-8 乙烯聚合的Green-Rooney機理
同位素標記研究認為,Green-Rooney機理需要進行必要的修正,中間體中有“抓氫鍵(agostic)”中間體化合物形成,被稱為“臨位抓氫鍵參與的乙烯聚合機理”(圖5-9)[19]。

圖 5-9 臨位抓氫鍵參與的乙烯聚合機理
以上研究主要集中在鈦催化劑催化乙烯聚合的方面,源自典型Ziegler催化劑的研究范疇,但是作者無意忽視Natta催化劑使用鈦催化劑催化丙烯聚合而獲得全同立構的聚丙烯,提高了所得產品聚烯烴的固化速率和強度。在Natta催化劑催化丙烯聚合之前,使用鉻系催化劑催化丙烯聚合,獲得了無規聚丙烯產品,因材料很難固化而沒有受到重視。在丙烯參與聚合時,由于多了一個甲基在雙鍵上,會有1,2-插入和2,1-插入兩種聚合的位置選擇性插入聚合方式,考慮到甲基的立體選擇性問題,會產生四種結構有所差異的聚丙烯產物,如圖5-10所示。
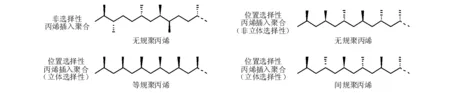
圖 5-10 丙烯插入聚合的三種方式與所得結構差異的四種聚丙烯
前面提過四價鈦和三價鈦的氯化物鹽用于乙烯聚合和丙烯聚合的區別,而進行催化劑制備的技術人員通常采用催化劑的顏色判斷鈦的價態:四價鈦為白色,負載催化劑固體呈現淺色;三價鈦具有暗紫色,其負載催化劑呈現較重的顏色。已經指出過,Natta的丙烯聚合催化劑認為是由“Et2AlCl-TiCl3”紅色催化體系組成,在最初十多年聚丙烯生產所需催化劑制備過程中,人們已經習慣使用氫氧化鎂和四氯化鈦在有乙醇存在下進行反應制備氧化鎂負載的三價鈦的氯化物,該反應過程中四價鈦顯然被乙醇還原形成了三價鈦化合物。早期的負載催化劑研究,無論乙烯聚合還是丙烯聚合催化劑,都能夠看到金屬氯化載體對于催化聚合活性的良好貢獻。期間,日本三井化學和三菱化學都在與德國和意大利聯合開發新的負載催化劑,使用了氯化鎂負載鈦的催化劑體系,使得催化聚合活性有了質的飛躍。此前的催化聚合所得聚烯烴中會有催化劑殘留,鈦殘留量高于30×10-6時造成聚烯烴樹脂有色,氯殘留量高于30×10-6時容易降解和腐蝕;因而,第二代Ziegler-Natta催化劑催化聚合所得聚烯烴通常需要洗掉無機鹽灰分,該過程通常稱為“脫色處理”工序。當氯化鎂負載催化劑工業化時,所得聚烯烴樹脂中鈦和氯的殘留量都低于30×10-6,在聚合工藝中免除了“脫色處理”工序;因而,在20世紀60年代末和70年代初研究的這類催化劑被稱為“第三代Ziegler-Natta催化劑”, 如對丙烯的聚合活性達到600 kg PP·(g Ti)-1,等規度達到98%[20-21]。
事實上,20世紀70年代后,聚烯烴Ziegler-Natta催化劑的活性提高非常有限,已不是研究的重點。燕山公司引進的我國首套聚烯烴生產的工業裝置是日本三井化學的工藝與設備,使用第三代Ziegler-Natta催化劑。針對這個催化劑體系,當時中國科學院化學所和隸屬于化工部的北京化工研究院以及石油部的石油科學研究院聯合攻關,獲得具有實用價值的聚丙烯催化劑,基于該技術分化和扶持了我國三家重要的聚烯烴催化劑生產廠家,分別是奧達催化劑公司、營口向陽化工廠和燕化高新催化劑公司;在國家部委整合情況下,燕化高新催化劑公司并入中國石化屬下的奧達催化劑公司,成為國際上頗具影響力和技術領先的聚烯烴催化劑生產企業。此后的40年里,人們把最初重視催化劑活性的研究轉到集中和關注所得聚合物樹脂的性能。針對聚烯烴樹脂的高性能,兩個有效途徑就是烯烴共聚和立體可控聚合;烯烴共聚的問題異常復雜,我們將在茂金屬鋯系催化劑部分進行涉及,立體可控聚合對于丙烯聚合更具有實質意義,后續的研究形成了第四代Ziegler-Natta催化劑。在第四代Ziegler-Natta催化劑中,少數催化劑改進通過載體控制實現[22],更多注意力是投到了伴隨鈦負載催化劑使用的脂類或者醚類化合物,其作用認為是可以調節催化活性中心的電子效應,被稱為“給電子體”[23]。基于這類“給電子體”加入到催化劑體系順序差異,制備載體催化劑時已經引入的脂或醚被稱為“內給電子體”,而在催化體系中采用與載體催化劑共混加入的這類脂或醚被稱為“外給電子體”。有時,會采用脂或醚同時做內給電子體和外給電子體,也會有多種脂或醚做給電子體;相關研究試探了大量的脂肪族和芳香族脂或醚以及二脂和二醚等。盡管有學者在發展給電子體催化劑體系中提出第五代Ziegler-Natta催化劑的建議,其效果和科學本質仍然是第四代催化劑的范疇。雖然難說Ziegler-Natta催化劑已經是“完美”的,但就催化活性而言,已達到每摩爾鈦催化劑每小時實現制備數千噸聚丙烯,全規聚丙烯選擇性超過98%的立體規整性。國內重要催化劑研究和生產廠家,中國石化奧達催化劑公司和營口向陽化工廠,都實現了第四代Ziegler-Natta催化劑的生產,在滿足國內聚烯烴生產的同時,出口到亞洲甚至歐美。
5.2.1.3鋯系(metallocene)催化劑
在討論鋯系催化劑時,不得不回到Ziegler-Natta催化劑的活性中間體研究探索的背景,進而逐漸演化成為鋯系(茂金屬)催化劑的發現歷史。發現Ziegler-Natta催化劑正值金屬有機化學學科的建立,聚烯烴催化學者力圖通過金屬有機化學的方法解釋催化機理和活性中間體[24],而金屬有機化學家則選擇具有重要應用價值的催化體系展示金屬有機的普遍性和實用價值[25],研究中一個交叉的重要體系就是二氯二茂鈦配合物與烷基鋁的催化體系(圖5-11)。表現出催化活性的助催化劑是二烷基氯化鋁,典型的是氯化二乙基鋁進行反應時形成二茂烷基化鈦氯化物,進一步與二烷基氯化鋁,形成二茂烷基鈦正離子催化活性中心[26-27];然而,當兩個烷基二茂鈦氯化物(如乙基二茂鈦氯化物)進行分子間β-氫轉移和形成一個乙烷分子脫去后,便產生乙基橋聯的氯化二茂鈦,該中間體沒有適合進行乙烯配位的空間,在進行歧化放出乙烯后形成氯化二茂鈦,三價鈦失去催化活性[28-29]。
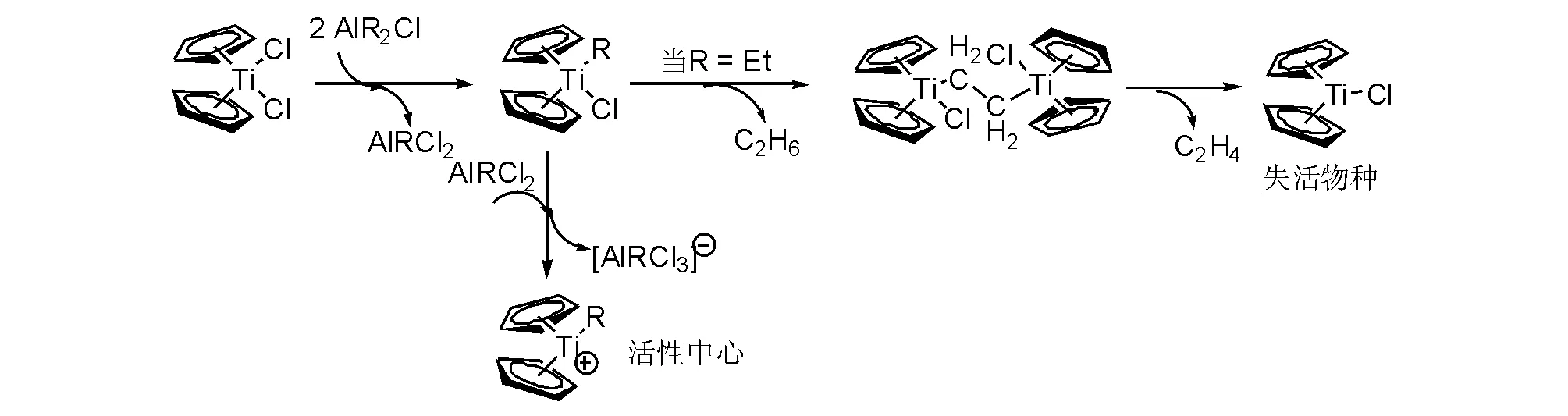
圖 5-11 茂金屬催化烯烴聚合的活性中間體與失活物種
這類乙基橋聯的氯化二茂金屬化合物,其鋯的化合物獲得了單晶,并測定了結構[30],證實乙基橋的每個碳原子同時聯到兩個鋯原子上。與此同時,為更好地使用核磁跟蹤催化中心的烷基化過程和研究催化聚合機理,而且為了更好地控制氫轉移的速率以便觀察茂基鈦的催化中間體,使用甲基比乙基更能夠簡化圖譜和便于研究。針對二甲基二茂鈦與三甲基鋁的反應進行跟蹤,成功地分離到甲基中α-氫轉移形成的亞甲基橋聯的茂基鈦化合物[31]。盡管證實該化合物中鈦的確以四價態形式存在,但是這類中間體并沒有呈現乙烯聚合活性;令人驚奇地發現,核磁管中凝結的水促進了乙烯聚合反應,并直接引入少量水在一升反應釜中再現了這類聚合催化[32]。該結果就是聚烯烴中具有里程碑意義的助催化劑甲基鋁氧烷(MAO),40年里,甲基鋁氧烷的結構問題一直吸引著研究者的注意,比較普遍接受的觀點就是不同聚集態的氧橋聯甲基鋁簇合物[33]。二茂鋯與三甲基鋁形成的體系對乙烯和丙烯都沒有催化性能,但二茂鋯與甲基鋁氧烷組成的催化體系卻比其同系物二茂鈦與甲基鋁氧烷的體系無論乙烯還是丙烯聚合都高出兩個數量級;不僅如此,茂基鋯化合物更容易制備和具有相對較好的溶解性,誘發了茂鋯催化劑的探索,引發了茂金屬催化烯烴聚合的集中研究與產業化發展,并成為近20年高性能聚烯烴樹脂研究與發展的推動力。
茂鋯配合物催化劑研究最多的是二茂鋯化合物的衍生物,并發展了不同橋聯的二茂鋯化合物,具有良好端烯烴聚合性能,具有代表性和不同結構對稱性模型的二茂鋯化合物見圖5-12。在合成這些二茂鋯配合物過程中,也合成了同族的鈦和鉿二茂金屬配合物,對同等條件下催化乙烯聚合性能進行比較[34],茂鋯配合物總是聚合活性較高。雖然茂鋯配合物的合成比茂鈦配合物容易,但前過渡金屬高價態的高親氧性還是增加了合成的難度,使得合成收率容易受到操作條件的影響。茂鋯配合物與Ziegler-Natta催化劑在催化乙烯聚合活性上同在一個數量級,Ziegler-Natta催化劑也可以通過聚合條件變化控制所得聚乙烯的分子量,盡管茂鋯配合物催化劑體系可以獲得聚乙烯的分子量分布窄,但客觀地講,在乙烯聚合應用上,茂鋯催化劑并不占任何優勢。
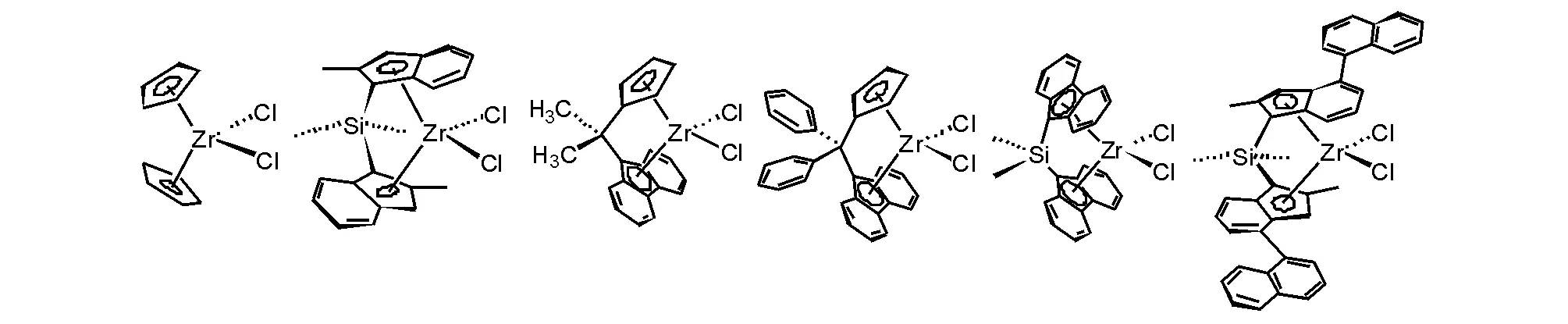
圖 5-12 二茂鋯催化劑的代表性模型
發揮茂金屬催化劑到極致的是其特效性實現可控聚合和共聚合。茂鋯催化劑用于α-烯烴的可控聚合能夠制備立體規整性不同和性能差異的聚烯烴樹脂,如聚丙烯樹脂材料(圖5-13)。
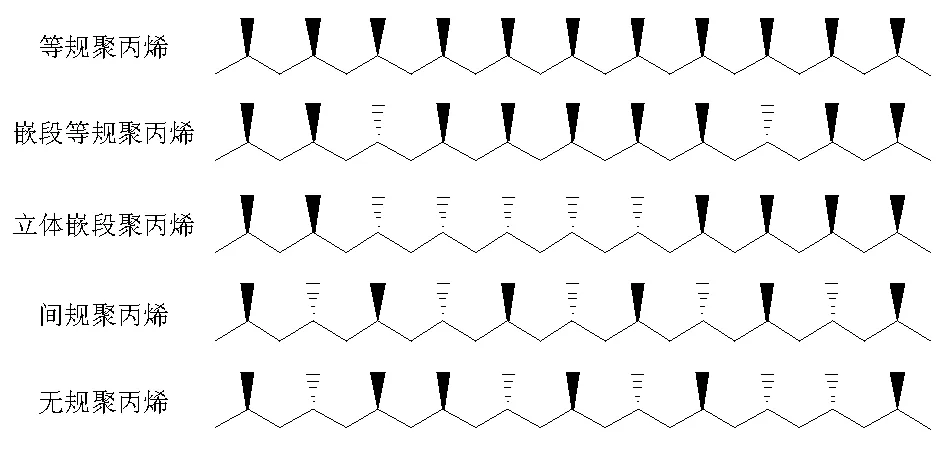
圖 5-13 聚丙烯的不同立體規整結構
聚丙烯材料中甲基的取向造成立體規整性差異,直接影響材料的結晶速率和結晶度以及密度和材料的物理與機械性能;在分子量相同的情況下,等規聚丙烯容易結晶成型,具有良好的耐熱性和機械強度,可以用于制造塑料制品和容器;無規聚丙烯耐熱性能差,結晶困難,呈現粘稠狀或蠟狀材料,在改性后使用,例如用于制備改性瀝青等。
茂鋯催化劑在聚烯烴中最具特殊地位,是催化乙烯與烯烴共聚最有效的催化劑體系,制備了品種繁多的新型高性能聚烯烴樹脂。針對聚烯烴樹脂材料性能提高進行的烯烴共聚研究是伴隨著烯烴自聚出現就誕生的研究,并且實現了釩系催化乙丙共聚膠催化體系仍有使用,Ziegler-Natta催化乙烯與α-烯烴(丁烯-1,己烯-1和辛烯-1等)的共聚是目前工業生產大品種線性低密度聚乙烯(LLDPE)的重要工藝。然而,釩系催化劑催化效率遠低于茂金屬催化劑體系,呈現茂金屬催化劑替代釩系催化劑的局面;在追逐更高機械性能和提高抗撕裂性能的材料研發中,需要提高乙烯與α-烯烴共聚樹脂中α-烯烴的共聚比例,不僅如此,還需要長鏈α-烯烴(如辛烯或者癸烯)的有效共聚,Ziegler-Natta催化劑就難于達到要求,需要茂金屬催化劑解決。還有一種特殊的功能聚烯烴樹脂,即乙烯與環烯烴(環戊烯或降冰片烯)共聚獲得工程塑料型聚烯烴樹脂,具有良好的透明性和抗撞擊性能。能夠實現共聚催化的茂金屬催化劑獲得學術界廣泛關注和研究,也有多個品種實現了產業化應用,但最值得知道和應用的高效茂金屬催化劑是硅橋聯四甲基茂基叔丁基胺化鈦二氯化物(圖5-14),即單茂橋聯鈦配合物催化劑,也被稱為“限定幾何構型的催化劑”(constrained geometry catalysts,CGC催化劑)。這個催化劑專利權最初歸陶氏化學(已過保護期),并與除中國外的世界聚烯烴大公司分享專利使用權,用于生產高性能聚烯烴,多數采用溶液聚合工藝,成為近20年來聚烯烴樹脂競爭發展的重要領域。由于國際聚烯烴對于高附加值市場的壟斷和我國聚烯烴
工程產業化能力差,我國還沒有規模化使用茂金屬催化劑制備高性能聚烯烴的裝置運行。
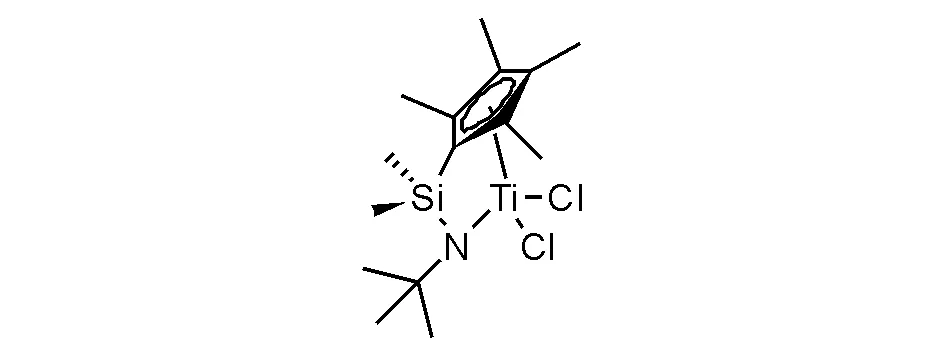
圖 5-14 限定幾何構型的催化劑
5.2.1.4后過渡金屬配合物催化劑
20世紀90年代中葉,α-二亞胺的鎳或鈀配合物(圖5-15,A)[35]和2,6-二亞胺吡啶的鐵或鈷配合物(圖5-15,B)[36-37]能夠實現乙烯聚合制備高分子量聚乙烯,由于后過渡金屬配合物制備簡單和穩定性高,很快吸引了學術界與產業界的廣泛關注,并獲得巨大的研究投入。圖5-15中A和B兩個模型是研究最多和獲得衍生物最為豐富,基于相同的理論基礎,發展了高效新型金屬配合物模型(圖5-15,C和D),呈現出更具實用價值的研究結果和新型聚合物材料。
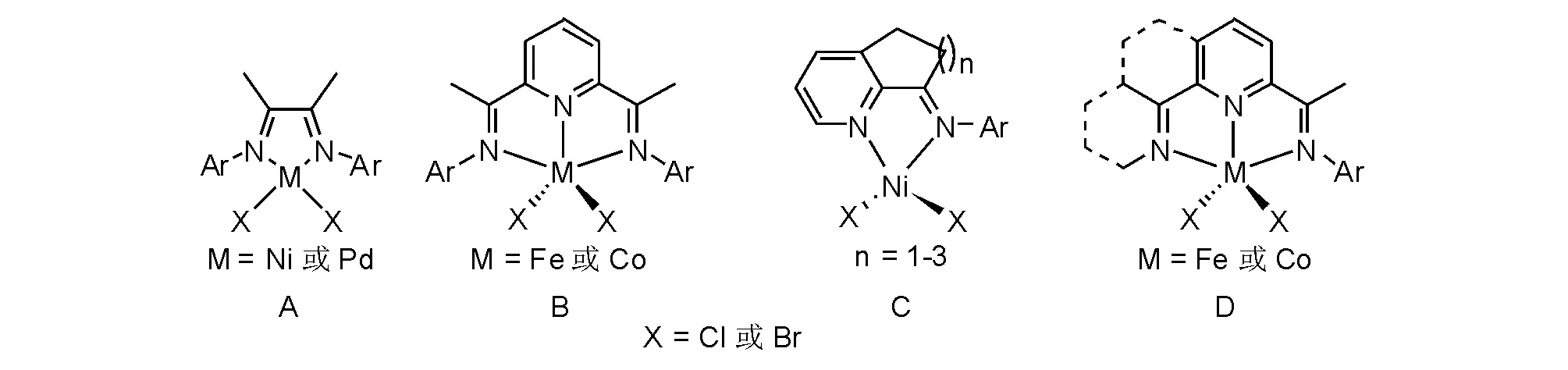
圖 5-15 典型后過渡金屬催化劑模型
20世紀70年代,第三代Ziegler-Natta催化劑在催化活性上已經滿足聚烯烴生產的需求,后續新型Ziegler-Natta催化劑和茂金屬催化劑都是基于所得聚烯烴品質開展研究和用于改進生產。后過渡金屬配合物催化烯烴聚合研究的突破口和意義,必然和應該集中在所得材料的性質與性能上。發現鎳和鈀配合物催化乙烯聚合過程中有更多氫轉移的過程,獲得了高度支化的聚乙烯甚至是聚乙烯彈性體[38];鐵和鈷配合物催化乙烯聚合獲得高度線性的聚合物,而且在分子量低的聚乙烯中能夠觀察到清晰的端雙鍵,成為制備不同高級α-烯烴的基石(作為新型長鏈共聚單體)和乙烯齊聚的高效催化劑[39]。相關的研究和產業化仍在進行中,所制備的新穎聚乙烯樹脂有可能影響到新穎高性能樹脂和材料的發展,幫助提高人們的生活水平和減少對環境的影響。
5.2.2合成橡膠材料及其催化聚合
在三大合成高分子材料中,橡膠占據了無可替代的地位,曾被作為軍用和國防物資受到各國政府的重視,國際橡膠統計網會不斷更新橡膠與彈性體的生產與應用。產量最大的三種合成橡膠依次分別是苯乙烯-丁二烯共聚物(styrene-butadiene rubber,SBR,簡稱為丁苯橡膠)年產三百多萬噸、順式-丁二烯橡膠(butadiene rubber, BR,順丁橡膠)年產兩百多萬噸和異戊橡膠(isoprene rubber, IR)年產接近一百五十萬噸,彌補了天然橡膠在產量和特殊性能需求上的不足。丁苯橡膠在1937年產業化,使用自由基聚合工藝,不在討論之列。聚丁二烯橡膠和異戊橡膠具有相似的催化聚合過程和工藝,在此,我們使用比較成熟的丁二烯聚合催化劑進行討論。
丁二烯聚合有兩種方式和三種結構規整的聚合物(見圖5-16),其中1,2-聚合所得聚合物含有大量的乙烯基,很容易交聯,不過這類聚合少,所得聚合物敏感,難有應用價值。1,4-聚合所得聚丁二烯又基于其結構趨向性獲得了反式聚丁二烯和順式聚丁二烯,基于聚合反應自身推動力,獲得順式聚丁二烯(順丁膠)結構聚合物更為普遍,其反式聚丁二烯認為是聚丁二烯產物中可以調節性能差異的組分,高順式含量的聚丁二烯是產業界努力追求的。
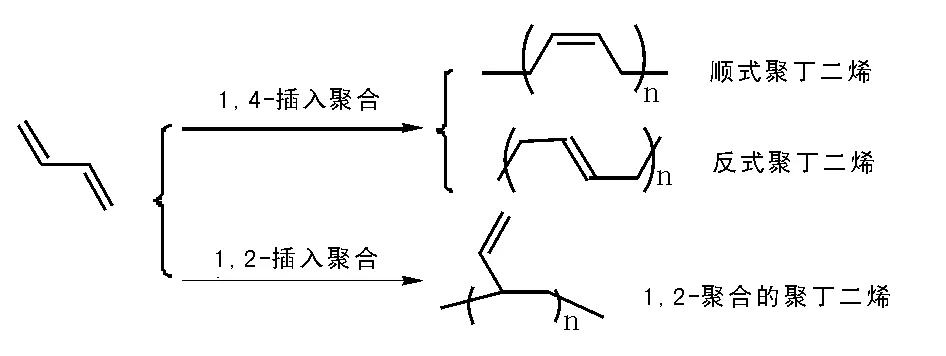
圖 5-16 丁二烯的聚合與所得聚合物
順丁橡膠是耐沖擊的熱塑性材料,具有優異的回彈性能和低的玻璃化溫度(Tg為-110 ℃),耐寒和耐磨性能好。與天然橡膠相比,其耐熱與抗老化性能更為優異,廣泛應用于輪胎、傳送帶、軟管卷涵蓋、鞋底和高爾夫球等,不僅如此,還廣泛用于其他熱塑性材料的改性,添加到高抗沖聚苯乙烯和高抗沖丙烯腈-苯乙烯-丁二烯共聚物(ABS樹脂)中,提高材料的性能,是重要的戰略物資。聚丁二烯主要采用溶液聚合工藝,通常使用鎳系、鈷系和釹系催化劑;其中,鎳系催化劑形成了成熟的催化體系,并獲得廣泛應用,鈷系催化劑高順式選擇性特點備受矚目,作為稀土大國更希望充分利用釹催化劑高活性和高選擇性獲得順丁橡膠生成的新突破。
工業上鎳系催化劑是三組分催化體系,主催化劑可以是乙酰丙酮鎳、水楊酸鎳、硬脂酸鎳和環烷酸鎳等;助催化劑由兩部分組成,其一是以三異丁基鋁、氫化二異丁基鋁或甲基鋁氧烷為主的烷基鋁化合物,其二是如三氟化硼為主的氟化物。此外,通常鎳系催化體系需要微量水參與陳化處理,鎳與鋁的比例一般為3~4,鋁與丁二烯比為0.23~0.35,水與丁二烯則接近等物質的量,這類催化劑配方是常用的催化劑體系。不難看出,鎳使用量較多,其催化活性提高是科學研究和技術發展的動力之一;文獻中有很多主催化劑研究的例子,Ai P等[40]研究的吡啶胺醇配合物模型(圖5-17)值得提及,其鎳配合物能夠室溫下可以引發聚合得到順式含量約為91%的聚丁二烯產品。

圖 5-17 新型金屬配合物丁二烯聚合催化劑
鈷系催化劑是兩組分催化體系,鈷鹽或者化合物為主催化劑,烷基鋁為助催化劑,能夠通過調整催化劑結構和控制聚合條件實現調控聚丁二烯產品的順式含量和結構選擇性,影響產物的宏觀性能。例如,使用甲基鋁氧烷助催化劑的體系中鹵化鈷和羧酸鈷所得聚合物為高順式聚丁二烯[41];然而,額外添加烷基膦或吡啶衍生物后,得到以1,2結構為主的聚丁二烯產物[42]。使用吡啶胺亞醇的鈷配合物(圖5-17),添加少量乙基倍半鋁(EASC),能夠對丁二烯實現順式選擇性的高效聚合;室溫下,2000倍的丁二烯能在1 h內完全轉化,聚合物的分子量為(2.31×105~3.08×105) g·mol-1,分子量分布為1.45~1.81,順式-1,4含量為96.3%~96.9%[43]。
稀土氧化物最先用于乙烯聚合[44],隨著研究深入,發現能夠更為有效地應用于二烯聚合,因而,部分學者將二烯聚合催化劑稱為Ziegler-Natta催化劑。 常用的釹催化體系由三組分構成,包括釹化合物主催化劑,還有烷基鋁化合物助催化劑以及活化劑;常見的助催化劑有三甲基鋁(TMA)、三乙基鋁(TEA)、三異丁基鋁(TIBA)、氫化二異丁基鋁(DIBAH)、甲基鋁氧烷(MAO)或者二丁基鎂(MgBu2)等,活化劑主要是乙基倍半鋁(EASC)、二氯化乙基鋁(DEAC)、氯化二乙基鋁、四氯化硅(SiCl4)和溴化鋁(AlBr3)等。最初的釹催化丁二烯聚合容易產生凝膠,凝膠的存在影響聚合物性能。經過大量研究和篩選,目前具有實用價值的釹催化劑多是羧酸釹化合物(圖5-18)[45]。
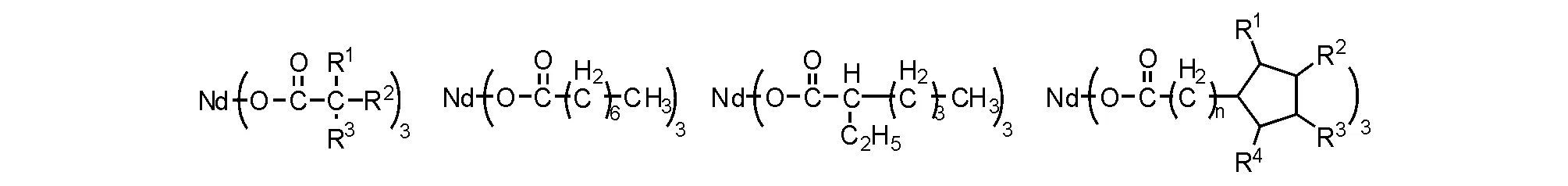
圖 5-18 常用的羧酸釹催化劑結構
依據使用主催化劑金屬的不同,又稱為鎳膠、鈷膠和稀土(釹)膠。目前,工業級順丁橡膠的主流產品還是鎳膠和鈷膠,其共同優點是催化劑活性高,產品中順式-1,4結構含量高,質量均勻,由此制得橡膠制品的綜合物理與機械性能較好。近十多年里快速發展的釹系催化劑獲得產業化發展,稀土順丁橡膠分子鏈擁有很高的規整度和線性結構,抗濕滑能力得到明顯改善。
5.2.3合成聚酯材料及其催化聚合
聚酯是由多元醇和多元羧酸縮聚所得,是廣泛應用的合成高分子材料,用于制備聚酯纖維和薄膜等。事實上,20世紀20年代DuPont公司和英國皇家化學工業公司(ICI)達成協議,共享特定合成材料的研究成果,因而,DuPont公司的研究成果很容易被英國學者獲得詳盡信息。基于DuPont公司的 Wallace Carothers博士在二酸與二胺反應制備具有里程碑意義的尼龍基礎上,1941年英國化學家John Rex Whinfield和James Tennant Dickson最先申請對苯二甲酸和乙二醇縮聚(圖5-19)制備聚對苯二甲酸乙二酯的專利,該催化技術于1946年在英國實現
產業化生產,1953年在世界范圍大規模工業化生產。在過去半個多世紀,雖然使用了多種二酸和二醇進行縮聚反應獲得高分子聚合物,其新材料的性能一直難于與已有的滌綸相媲美,因而,更多的研究與產業化集中在滌綸制備催化劑的研究;然而,抽象地考慮縮聚反應本身,就是縮合脫水過程和所得聚酯材料聚合度的控制問題。
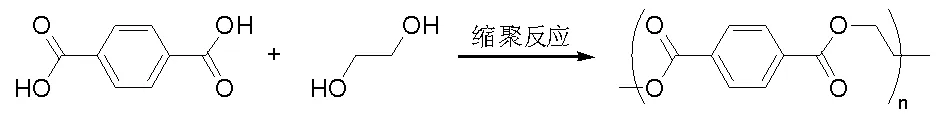
圖 5-19 苯二甲酸和乙二醇縮聚反應制備滌綸
目前,滌綸年產超過50 Mt,其中33 Mt用于制備聚酯纖維。滌綸合成乙二醇外,對苯二酸根的原料是對苯二甲酸或者對苯二甲酸二甲酯;對苯二甲酸二甲酯雖然在反應過程中會有甲醇副產物產生,但原料便于儲運和提純,是制備高品質滌綸所通用的。對于縮聚反應機理研究表明,該縮聚反應是通過酯化或脂交換反應,首先獲得苯二甲酸縮二乙二醇中間體,該中間體進一步進行縮合脫去一個乙二醇,形成聚對苯二甲酸乙二酯(圖5-20)。
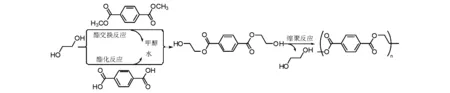
圖 5-20 滌綸合成機理
根據復合反應驅動力,只要能夠加快第一步酯交換,或者酯化反應脫出的甲醇,或者水被轉移與吸收,就比較容易地推動苯二甲酸縮二乙二醇中間體的形成;縮聚反應副產物是反應原料乙二醇,這步縮聚反應成了制備滌綸的控制步驟,其催化劑研究成為制備滌綸過程中研究的中心環節。滌綸生產所用的催化劑包括銻系、鍺系和鈦系三類,所得滌綸性能也略有差異。鍺系催化劑所得滌綸呈現白色,具有切片性能優異和結晶速率快的優點,但存在氧穩定性差和熱穩定性一般,鍺的價格昂貴,除了滿足光學材料和高透明材料需求外,難于大規模生產。鈦系催化劑所得滌綸只是結晶速率好,其色澤和熱氧穩定性都較差,但鈦的低毒性質還是幫助所得滌綸材料一定的應用。盡管銻的殘留可能導致白血病以及影響肝臟和脾臟的代謝,但是銻廉價和所得滌綸良好的結晶與熱氧穩定性,還是使這類聚酯獲得最為廣泛的應用,占據了聚酯生產的八成以上。除此之外,還有鋁系和錫系等催化劑體系在產業化研究和推進中。
乙二醇銻被認為是最為普遍應用的工業催化劑,其聚集態結構獲得了單晶X-光晶體衍射的確認(圖5-21)。其用于催化乙二醇與對苯二甲酸或者對苯二甲酸二甲酯進行縮聚制備滌綸過程中呈現銻離子外僅僅是乙二醇的存在,不會干擾反應物純度,實現穩定生產的效果。此發現后,以往的銻系催化劑(三氧化二銻或者醋酸銻)逐漸被替代掉;與此同時,不難理解以往使用的銻催化劑很可能在體系中自動轉化為乙二醇銻催化物種。
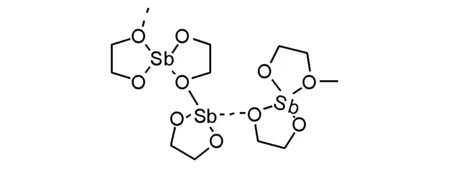
圖 5-21 乙二醇銻的結構
為了開發替代銻系催化劑的新型高效和低(無)毒催化劑體系,近些年比較活躍的研究課題是鈦系催化劑體系,主要還是使用鈦酸脂(如鈦酸丁酯或者鈦酸異丙脂等)以及鈦酸芳香脂和復合脂類化合物,力求提高催化聚合活性和降低鈦在聚酯中的殘留,改善聚酯的色度;不僅如此,還希望提高所得聚酯的分子量和降低分子量分布,以便提高聚酯的耐氧和耐熱穩定性。總之,聚酯生產了70年,其需求和生產的發展仍然持續,并期望在低毒聚酯生產技術上獲得突破。
5.3展望
高分子合成發展近一個世紀,使人們的居住、交通設施乃至食品包裝和保鮮都發生了極大的變革,使得人類社會逐漸擺脫了依賴天然材料和無機材料的歷史。在經歷探索和發展階段后,高分子材料的廣泛應用和普遍推廣滿足了對于“自然資源消耗減量,但社會繁榮和人們生活高水準”的要求,替代自然材料不僅保護了自然資源而且減少了防腐和成品變型的缺點,滿足了人們對于審美的要求,提高了成品的質量。隨著化學和物理學的認知發展,新材料研究和持續發展仍在進行中,其合成和制備工藝中減少副產物產生和高分子材料微結構控制都可以借助催化效率提高和高效催化劑的研制實現。在量大面廣的高分子合成中,任何合成與加工工序中微小的效率提高都將極大地提升產業的附加值。本章節列舉的只是三個大品種傳統高分子材料,并沒有介紹特種高分子材料和高分子專用料材料的催化問題。就文中涉及的聚烯烴催化問題,被稱為“塑料之王”的超高分子量聚烯烴材料其在北京生產所用催化劑僅是在普遍使用的Ziegler-Natta催化劑基礎上條件控制實現的,這種條件控制卻喪失了產量效益,只是常規聚烯烴生產量的三分之一;盡管目前超高分子量聚乙烯每年只有數十萬噸的需求,一旦新型高效催化劑出現降低了生產成本,在合理價格基礎上肯定會有飛躍性應用發展。常規聚烯烴生產的催化劑雖然成熟,聚烯烴新材料特別是后過渡金屬催化劑制備的聚乙烯新材料還剛剛起步;無論高度線性聚乙烯材料還是高度支化聚乙烯材料都是以往產業界所不能預測的新材料,高度支化聚乙烯材料一旦可以添加部分二烯共聚單體,就可能引發橡膠新材料的革命。目前二烯催化合成橡膠的催化效率還普遍比較低,催化劑在橡膠及其制品中的殘留值都很高,硫化交聯后仍有很大部分雙鍵成為橡膠制品老化的推進劑;橡膠合成的高效催化劑具有迫切需要。盡管銻的毒副作用獲得普遍關注,銻系聚酯中每克聚酯的銻殘留仍為(120~350) μg,而且這類聚酯呈現淺灰色;這種色澤的缺點與快速發展的聚酯市場形成巨大的差異,更難于滿足人們對于降低金屬(毒副作用)殘留的需求,必將需要一場聚酯催化劑的新革命。
不僅如此,合成高分子材料使用后的有效利用和污染控制問題形成了另一個方興未艾的催化課題:高分子材料降解成為有機小分子,甚至是高分子合成的單體;這將有利于形成合成高分子材料“制備-使用-處理”的良性循環。合成,陽光下的上帝之路;降解,夜幕中的物能之智。師從自然,人為加速;自然界植物不斷進行光合作用提供材料,人類與動物不斷消耗和分解,形成完美循環。催化角度看問題,數千年前人們就知道了“釀酒”和“發酵”的菌種這類生物催化劑。目前,出現了高分子材料生物降解和催化降解兩個有效途徑,在“時間就是效率”的背景下,需要篩選出高效和快速的降解途徑,化學工程的熱裂解和催化降解除了仍需不斷找尋高效催化劑外,制約技術的瓶頸還是高分子材料的回收和分類集中的問題。
總之,合成高分子材料以及其催化劑與工藝的研究發展是目前和未來半個世紀材料科學革命的基礎,而材料革新必將促進交通運輸工具和工程技術的飛躍發展。無論從專業角度還是社會需求,都為合成高分子材料相關的催化問題研究提供了廣闊的空間,并為社會和每個參與者提供了光明的未來。
參考文獻:
[1]Goodyear C.Improvement in India-rubber fabrics:US,3633[P].1844-06-15.
[2]BrydsonJ A.Plastics materials[M].6th Edition,Oxford:Butterworth-Heinemann,1995.
[3]Baekeland L H.Chemical achievers:the human face of chemical sciences,chemical heritage foundation(2005)[M].Retrieved.2007-11-08.
[4]Fawcett E W.The formation of high polymers[J].Journal of the Chemical Society,1936,32:119-121.
[5]Hogan J P,Banks R L.Polymerization of olefins:US,2825721[P].1954-08-21.
[6]Clark A.Polymerization and copolymerization of liefins on chromium oxide catalysts[L].Advances in Chemical Physics,1969,91:387-397.
[7]Ruddick V J,Badyal J P S.CO reduction of calcined CrOx/SiO2ethene polymerization catalysts[J].Langmuir,1997,13:469-472.
[8]McDaniel M P,Collins K S,Benham E A,et al.The activation of Phillips Cr/silica catalysts Ⅴ.Stability of Cr(Ⅵ)[J].Applied Catalysis A:General,2008,335:252-261.
[9]Cossee P.Ziegler-Natta catalysisⅠ.Mechanism of polymerization of alpha-olefins with Ziegler-Natta catalysts[J].Journal of Catalysis,1964,3:80-88.
[10]Gr?neveld C,Wittgen P P M M,Swinnen H P M,et al.Hydrogenation of olefins and polymerization of ethylene over chromium-oxide silica catalystsⅤ.In situ infrared measurements and investigation of the polymer[J].Journal of Catalysis,1983,83:346-361.
[11]Ellermann V J,Hagen K,Krauss H L.Chemistry of polyfunctional ligands:complexes of chromium(Ⅱ)-chloride and surface chromium(Ⅱ) with polydentatep-ligands and as-ligands[J].Zeitschrift Fur Anorganische Und Allgemeine Chemie,1982,487:130-140.
[12]McDaniel M P.Supported chromium catalysts for ethylene polymerization[J].Advances in Catalysis,1985,33:47-98.
[13]Ziegler K,Holzkamp E,Martin H.Polymerisation vonthylen und anderen olefinen[J].Angewandte Chemie,1955,67:426-426.
[14]Ziegler K,Holzkamp E,Martin H.Das mülheimer normaldruck-poly?thylen-verfahren[J].Angewandte Chemie,1955,67:541-547.
[15]Natta G,Pino P,Corradini P,et al.Crystalline high polymers of alpha-olefins[J].Journal of the American Chemical Society,1955,77:1708-1710.
[16]Boor J.Ziegler-Natta catalyst and polymerization[M].New York:Academic Press,1979.
[17]Ivin K J,Rooney J J,Stewart C D,et al.Mechanism for stereospecific polymerization of olefins by Ziegler-Natta catalysts[J].Journal of the Chemical Society,1978,14:604-606.
[18]Green M L H.Studies on synthesis,mechanism reactivity of some organo-molybdenum and organo-tungsten compounds[J].Pure and Applied Chemistry,1978,50:27-35.
[19]Piers W E,Bercaw J E.Alpha-agostic assistance in Ziegler-Natta polymerization of olefins-deuterium isotopic perturbation of stereochemistry indicating coordination of an alpha-C—H bond in chain propagation[J].Journal of the American Chemical Society,1990,112:9406-9407.
[20]Galli P,Barbe P C,Noristi L.High-yield catalysts in olefin polymeriztion-general outlook on theoretical aspects and industrial uses[J].Angewandte Makromolekulare Chemie,1984,120:73-90.
[21]Galli P,Luciani L,Cecchin G.Advances in the polymerization of polyolefins with coordination catalysts[J].Angewandte Makromolekulare Chemie,1981,94:63-89.
[22]Galli P,Milani F,Simonazzi T.New trends in the field of propylene based polymers[J].Polymer Journal,1985,17:37-55.
[23]Busico V.Giulio Natta and development of stereoselective propene polymerization[J].Advances in Chemical Physics,2013,257:37-57.
[24]Natta G,Pino P,Mazzanti G,et al.Polimerizzazione stereospecifica delle alfa-olefine Nota[J].Gazzetta chimica Italiana,1957,87:549.
[25]Breslow D S,Newburg N R.Bis-(cyclopentadienyl)-titanium dichloride-alkylaluminum complexes as catalysts for the polymerization of ethylene[J].Journal of the American Chemical Society,1957,79:5072-5073.
[26]Chien J C W.Kinetics of ethylene polymerization catalyzed by bis-(cyclopentadienyl)-titanium dichloride dimethylaluminum chloride[J].Journal of the American Chemical Society,1959,81:86-92.
[27]Waters J A,Mortimer G A.Study of (pi-C5H5)2Ti(C2H5)Cl and its higher homologs in soluble Ziegler catalysts[J].Journal of Applied Polymer Science,1972,10:895-907.
[28]Sinn H,Patat F.Uber die wirkungsweise metallorganischer katalysatoren[J].Angewandte Chemie,1963,75:805-813.
[29]Sinn H,Bandermann F,Hinck H A.Polymerization reaction of trimethylaluminum [J].Angewandte Chemie International Edition,1968,7:399-414.
[30]Kaminsky W,Kopf J,Sinn H,et al.Extreme bond angle distortion in organozirconium compounds active toward ethylene[J].Angewandte Chemie International Edition,1976,15:629-630.
[31]Heins E,Hinck H,Kaminsky W,et al.Examples of condensation reactions of emthyl alkyls with cleavage of alkanes[J].Makromol Chem,1970,134:125.
[32]Andresen A,Cordes H G,Herwig J,et al.Halogen-free soluble Ziegler catalysts for polymerization of ethylene-control of molecular-weight by choice of temperaure[J].Angewandte Chemie International Edition,1976,15:630-632.
[33]Glaser R,Sun X.Thermochemistry of the initial steps of methylaluminoxane formation.Sluminoxanes and cycloaluminoxanes by methane elimination from dimethylalumium hydroxide and its dimeric aggregates[J].Journal of the American Chemical Society,2011,133:13323-13336.
[34]Kaminsky W.New polymers by metallocene catalysis[J].Macromolecular Chemistry and Physics,1996,197:3907-3945.
[35]Johnson L K,Killian C M,Brookhart M.New Pd(Ⅱ)-based and Ni(Ⅱ)-based catalysts for polymerization of ethylene and alpha-olefins[J].Journal of the American Chemical Society,1995,117:6414 -6415.
[36]Small B L,Brookhart M.Highly active iron and cobalt catalysts for the polymerization of ethylene[J].Journal of the American Chemical Society,1998,120:4049-4050.
[37]Britovsek G J P,Gibson V C,Kimberley B S,et al.Novel olefin polymerization catalysts based on iron and cobalt[J].Chemical Communications,1998,63:849-850.
[38]Wang S,Sun W H,Redshaw C.Recent progress on nickel-based systems for ethylene oligo-/polymerization catalysis[J].Journal of Organometallic Chemistry,2014,751:717-741.
[39]Ma J,Feng C,Wang S,et al.Bi- and tri-dentate imino-based iron and cobalt pri-catalysts for ethylene oligo-/polymerization[J].Inorganic Chemistry Frontier,2014,1:14-34.
[40]Ai P,Chen L,Guo Y,et al.Polymerization of 1,3-butadiene catalyzed by cobalt(Ⅱ) and nickel(Ⅱ) complexes bearing imino- or amino-pyridyl alcohol ligands in combination with ethylaluminum sesquichloride[J].Journal of Organometallic Chemistry,2012,705:51-58.
[41]Nath D C D,Shiono T,Ikeda T.Cis-specific living polymerization of 1,3-butadiene with CoCl2and methylaluminoxane[J].Macromolecular Chemistry and Physics,2002,203:756-760.
[42]Ricci G,Sommazzi A,Masi F,et al.Well-defined transition metal complexes with phosphorus and nitrogen ligands for 1,3-dienes polymerization[J].Coordination Chemistry Reviews,2010,254:661-676.
[43]Jie S,Ai P,Li B G.Highly active and stereospecific polymerization of 1,3-butadiene catalyzed by dinuclear cobalt(Ⅱ) complexes bearing 3-aryliminomethyl-2-hydroxybenzaldegydes[J].Dalton Trans,2011,40:10975-10982.
[44]Brown C P,Saunders J.Polymerization Catalysts for ethyleneⅠ.Rare earth oxides [J].Journal of Polymer Science,1960,43(142):579-579.
[45]Friebe L,Nuyken O,Obrecht W.Neodymium-based Ziegler/Natta catalysts and their application in diene polymerization[J].Advances in Polymer Science,2006,204:1-154.
孫文華,1963年生,博士,無黨派民主人士,中國科學院化學研究所二級研究員和中國科學院大學崗位教授。
1986年在蘭州大學完成本科學習后,1986-1989年在中國科學院蘭州化學物理研究所完成碩士學位并留所工作,1991-1994年在中國科學院蘭州化學物理研究所完成在職博士學習;同期,1993年12月經中國科學院批準在中國科學院蘭州化學物理研究所任副研究員。1995年11月-1999年10月在日本學術振興會、日本科學技術事業團和日本文部省外國人客座教授資助下在日本北海道大學催化中心工作;1999年10月在中國科學院“百人計劃”支持下到中國科學院化學研究所任研究員。其蘭州工作集中在羰基原子簇設計合成和氫甲酰化催化,日本工作集中在有機合成方法學。此外,作為洪堡基金會訪問教授在德國明斯特大學、日本文部省訪問教授在名古屋大學、以及法國路易斯-帕斯卡大學和斯特拉斯堡大學訪問教授,進行訪問研究。在中國科學院化學研究所的工作集中開展“過渡金屬配合物烯烴聚合”催化劑與聚合工藝,以及新型聚烯烴材料研究;發表研究論文300余篇,獲得授權專利57件,是愛思唯爾統計2014年和2015年“中國高被引化學研究者”之一。基于其研究成果,獲得了“2009年度北京市科技進步一等獎”,“2011年中國石油和化學工業聯合會科技進步獎一等獎”,2011年當選為英國皇家化學會會士,2012年獲得中國僑界創新人才貢獻獎,2013年獲得第七屆馮新德高分子獎最佳文章獎。
現代催化化學講座
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
