典型聚合物中多種碳的化學位移量化計算
2016-05-21 08:53:15付維貴劉珊珊費睦融天津工業大學膜分離與膜過程省部共建國家重點實驗室天津300387
天津工業大學學報 2016年2期
付維貴,潘 靖,劉珊珊,費睦融,陳 莉(天津工業大學膜分離與膜過程省部共建國家重點實驗室,天津300387)
?
典型聚合物中多種碳的化學位移量化計算
付維貴,潘靖,劉珊珊,費睦融,陳莉
(天津工業大學膜分離與膜過程省部共建國家重點實驗室,天津300387)
摘要:使用Gaussian09量化計算軟件,基于密度泛函理論(DFT),采用B3LYP//GIAO方法基于6-311G(2d,p)基組,計算聚合物聚苯醚(PPO)、聚N-異丙基丙烯酰(PNIPA)中多種碳的13C NMR化學位移.通過與實驗值進行比較,準確歸屬不同環境下的碳的化學位移,并得到分子結構、構象等因素對化學環境的影響.
關鍵詞:量子化學計算;核磁共振;化學位移;密度泛函理論
量子化學(quantum chemistry)是應用量子力學的基本原理和方法研究化學問題的分支學科[1-2],它是化學、物理、材料科學研究的重要工具,不僅可以預測化合物的核磁性質、熱力學性質、過渡態能量和結構、紅外和拉曼光譜等,還可以了解生物大分子某活性位點的電荷轉移.其中,核磁共振譜圖的化學位移包含分子體系中的重要信息:一是分子結構的信息;二是分子間相互作用信息[3-4].原子核的化學位移由其所處的分子環境確定,各種影響電子云密度的因素都將影響化學位移,其中各向異性、電負性效應及氫鍵相互作用等對化學位移影響最大.化學位移譜峰的歸屬僅是通過實驗的手段進行測量分析,對于一些復雜化學結構的確認,會有一些困難.引入理論計算方法進行預測,可以提高化學位移歸屬的準確性[5-6],即用高斯軟件(如Gaussian09),運用數值方法預測分子的性質[7-9].徐璐[10]將固體核磁共振實驗結果與量子化學計算相結合,在不同的初始構型、構象及不同優化方式條件下,得出計算結晶性高分子核磁共振化學位移的可行性方法. Fu等[11]對PMMA和PVPh共混體系進行研究,計算得到與羰基形成氫鍵相互作用的羥基的化學位移值,理論計算結果與多脈沖固體1H NMR實驗譜擬合結果一致.本文利用Gaussian 09量化計算軟件,獲取PPO和PNIPA兩種典型的聚合物的化學位移理論值,通過與實驗值比對,進一步對譜峰進行準確歸屬,解釋同種碳在不同化學環境下的化學位移變化及譜峰劈裂的原因.
1 典型聚合物PPO和PNIPA的多種碳
聚苯醚(PPO)分子中含有大量的芳香環結構,每個苯環上有2個對稱的次甲基(CH),但固體13C NMR譜中,譜峰存在劈裂且分辨率降低,不易區分.
聚N-異丙基丙烯酰胺(PNIPA)的結構式中含有較大的側基,主鏈和側基上都存在不同環境下的烷基碳(CH,CH2),對應的實驗譜圖存在譜峰疊加,分辨率低,不易區分.
以上2種聚合物體系中,不同環境下的多種碳的化學位移明顯不同,存在或譜峰疊加不易分辨等現象,本文采用量化計算的方法得到理論化學位移值,與實驗譜圖相結合,對譜峰進行有效的歸屬.
2 量化計算方法
高斯量化計算均在Linux操作系統下,采用四核并行計算.運用Gaussian 09量化計算軟件,基于密度泛函(DFT)理論,采用B3LYP // 6-311G +(2d,p)基組進行結構優化;并在此基礎上,選用較為經典的Gauge-Including Atomic Orbital(GIAO)方法化進行NMR化學位移計算[12-13],化學位移(δ)單位為10-6,以四甲基硅烷(TMS)的13C NMR理論計算值為參考.實驗數據和理論計算的譜圖,均在Origin軟件中進行分析處理.
3 結果與討論
3.1 PPO中多種碳的化學位移
搭建PPO二聚體,優化后的模型圖如圖1所示.
計算得到的PPO化學位移的理論譜圖與實驗譜圖[14]進行比對分析,如圖2所示.圖2中曲線是實驗譜圖,下面的豎線表示量化計算所得化學位移理論值.實驗譜峰擬合值和13C NMR譜的理論化學位移值,列于表1中.
通過表1中實驗值和理論值對比可以看出,各種碳的化學位移理論值和實驗值幾乎相近,說明結構搭建及計算結果合理有效.

表1 PPO13C NMR實驗值和理論化學位移計算值Tab.113C NMR experimental value and theoretical calculatedvalue of chemical shifts for PPO 10-6
對比苯環上不同位置上的次甲基(CH)的化學位移,其中CH(C2)譜峰劈裂;而季碳C1和C5的譜峰明顯分開. C2在分子結構式中雖然處在對稱的位置上,但在實際分子構象中(圖1),一個遠離甲基(如21號碳原子距離11號質子0.426 nm),一個靠近甲基(如22號碳原子距離11號質子0.316 nm),化學環境發生變化,從而出現譜峰劈裂,化學位移理論值分別為119.25×10-6和115.01×10-6,各自與相應的實驗曲線譜峰擬合結果相接近.遠離CH3(靠近端羥基)的C(C1)周圍電子云密度小,化學位移處在高頻處(160.00× 10-6);而遠離端羥基與醚氧鍵—(C—O—C)—鍵連的C(C5)靠近2個CH3,周圍電子云密度大,因此在低頻處(150.55×10-6).從而說明,該分子中多種碳局域化學環境的不同與分子構象相關.
3.2 PNIPA中多種碳的化學位移
搭建PNIPA三聚體與NIPA單體相互靠近的模型,模擬聚合物分子間氫鍵的形成,結構優化模型如圖3所示.圖3中虛線鏈接的代表三聚體中C=O氧原子(原子標號38)與單體側基上的NH質子(原子標號88)間形成一對氫鍵.將計算得到的理論值與文獻[14]中的實驗譜圖進行比對分析,如圖4所示.圖4中曲線為實驗譜圖,下面的豎線是理論計算值.實驗譜圖擬合得到的峰值和13C NMR理論化學位移值列于表2中.
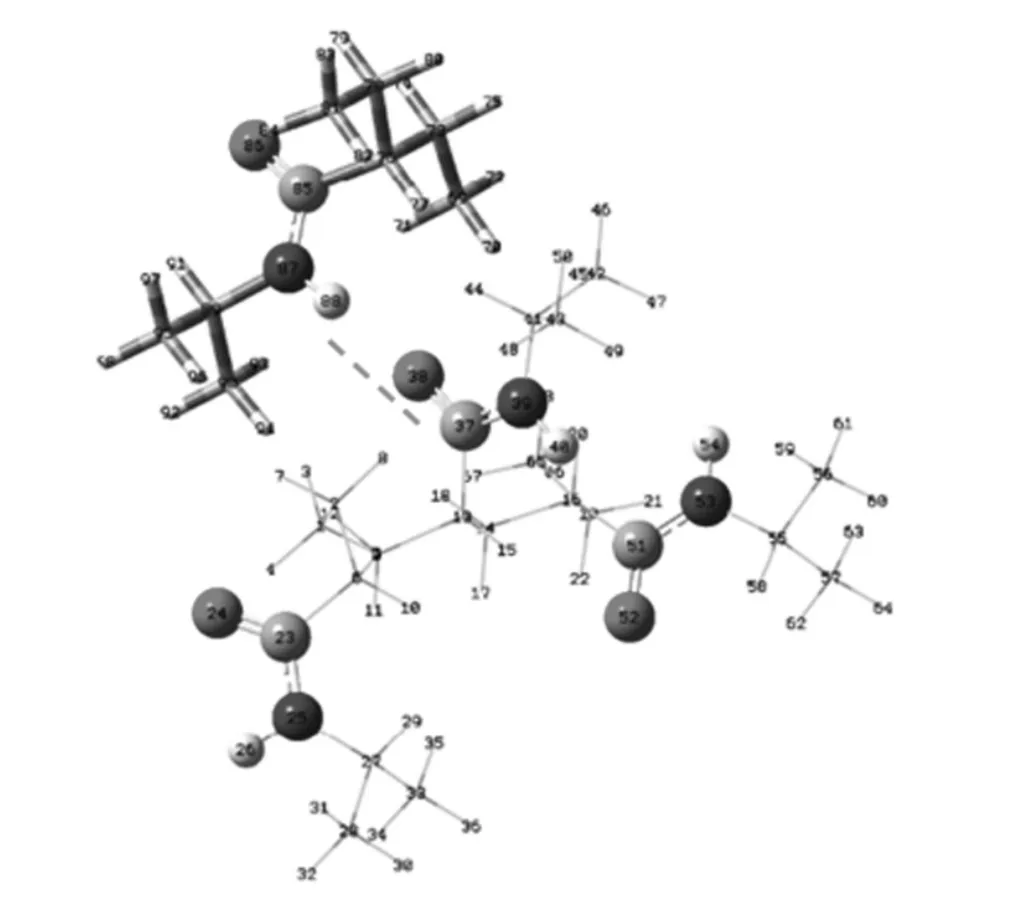
圖3 PNIPA三聚體與單體形成分子間氫鍵優化結構模型圖Fig.3 Optimized hydrogen-bond configuration between PNIPA trimer and monomer
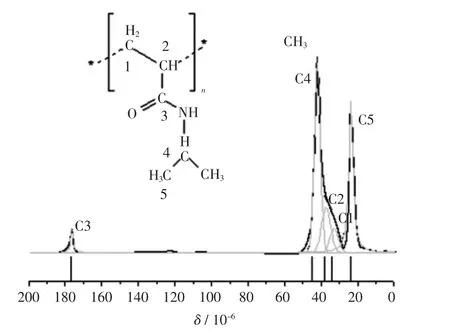
圖4 PNIPA結構式、13C NMR實驗譜和化學位移理論計算譜Fig. 4 Molecular structure(top),experimental 13C NMR spectra(medium)and calculated chemical(bottom)of PNIPA
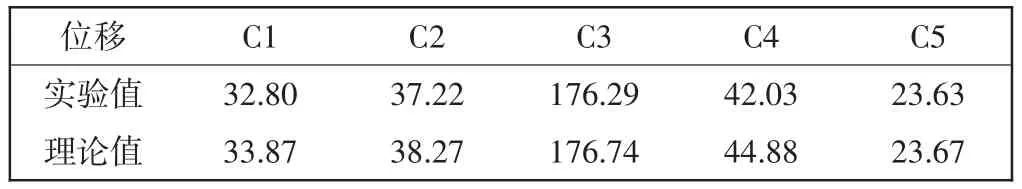
表2 PNIPA13C NMR實驗值和理論化學位移計算值Tab. 213C NMR chemical shifts of PNIPA by experimentation and theoretical calculation 10-6
從圖4中實驗譜圖分峰擬合結果可以看到,主鏈上的CH(C2)13C NMR化學位移在37.22×10-6附近,而側基上CH(C4)的化學位移為42.03×10-6;從表2中對應的理論值可以看到,CH(C2)的化學位移為38.27× 10-6,側基上CH(C4)的化學位移在44.88×10-6附近.當側基上的羰基(C=O)上的氧原子(原子標號為38號)與另一個單體分子側基上的氨基(NH)質子(原子標號88)形成分子間氫鍵后(如圖3所示),與羰基碳鍵連的主鏈上的CH上的碳原子(原子標號13)的化學位移較形成氫鍵前向低頻方向移動了約1×10-6.這里實驗值與理論計算值略有偏差,一方面是由于實驗值是各種構象加權的結果,同時分峰擬合值也會有人為誤差;另一方面,PNIPA分子側基較大,空間位阻相應較大,多條分子鏈間形成多氫鍵體系時,在不同位置上的同種碳的化學位移也會由于空間構象的變化而不同,這里我們用簡單的模型優化計算結果區別不同的烷基碳,定性的幫助對實驗譜圖的譜峰歸屬.實驗譜峰擬合和量化計算結果絕對值的比較,還有待于更多單元和多條分子鏈分子間氫鍵的模擬結果.
從PNIPA結構式上看,主鏈上的CH2(C1)和CH (C2),及側基上CH(C4)的譜峰疊加,由于主鏈上的CH (C2)與側基上的羰基(C=O)相連,去屏蔽效應增加,因此C2相對于C1(實驗擬合值δ= 32.80×10-6)略向低場處偏移.而側基上的CH(C4)與氨基(NH)鍵連,使得電負性增加,周圍電子云密度降低,相應化學位移向高頻處移動(>42×10-6),因此高頻處(42.03×10-6)的尖峰歸屬為PNIPA側基上的CH的信號峰.該理論計算結果與固體核磁共振實驗中采用魔角旋轉下極化反轉自旋交換(PISEMA)方法對CHn信號的鑒別結果是一致的,從42×10-6左右的峰的偶極劈裂譜線型可以判斷該處只有CH信號,沒有主鏈上CH2的偶極劈裂線型出現[14].固體狀態下,由于偶極耦合作用的影響會使譜峰展寬互相疊加,用量化計算方法計算中心位移值,對幫助固體或液體核磁共振譜圖解析是有效的,本文對主鏈上CH和側基CH的計算結果與文獻[14]中的實驗歸屬一致,與文獻[15]中的液體核磁共振譜圖的歸屬還存在分歧.
此外,還搭建了由多個單體組成具有不同立構的分子模型,結構優化及計算結果表明,立構規整度對主鏈上CH和CH2的化學位移影響較大,化學位移偏移范圍可達5×10-6~10×10-6,因此實驗譜圖擬合結果顯示這2種基團譜峰較寬,而對側基上CH的影響不大(偏移在2×10-6范圍內).
4 結論
本文使用Gaussian 09量化計算軟件,基于密度泛函(DFT)理論,采用B3LYP // 6-311G +(2d,p)方法,分別對PPO和PNIPA聚合物中多種碳進行13C NMR化學位移理論計算,并與實驗譜圖相比較.研究表明:
(1)對于典型聚合物中多種碳的核磁共振譜圖的化學位移,由于誘導效應、分子間氫鍵等的影響,分子構象的不同,使原子周圍化學環境發生變化,導致化學位移發生變化或譜峰劈裂.
(2)通過量化計算方法,可以幫助復雜體系的核磁共振等實驗譜圖的解析和譜峰歸屬.
參考文獻:
[1] RITA C G,LEIF A E. Theoretical study of hypericin[J]. J Photoch Photobio A,2005,172(3):293-299.
[2] MARCIN H,JACEK R. Effects of substituting a OH group by a F atomin D glucose:Ab initio and DFT Analysis [J]. J Am Chem Soc,2001,123(10):2308-2316.
[3] ERNST R R,BODENHAUSEN G,WOKAUN A. Principles of Nuclear Magnetic Resonance in One and Two Dimensions[M]. Oxford:Oxford University Press,1991.
[4] SCHMIDT-ROHR K,SPIESS H W. Multidimensional solidstate NMRandpolymers[M].Pittsburgh:Academic Press,1994:402-439.
[5] ANETA J,JAROSLAW P,STANISLAW R. DFT study of a novel lead structure in the isoxazole heterocyclic system [J]. J Mol Struct,2003,636(1/2/3):203-214.
[6] MACIEJ P,JERZY K,MIROSLAW P,et al.1H,13C NMR studies and GIAO/DFT calculations of substituted N-(4-aryl-1-piperazinyl- butyl)derivatives,new analogues of buspirone [J]. J Mol Struct,2004,698(1/2/3):93-102.
[7] HELGAKER T,JASUNSKI M,RUUD K. Ab initio methods for the calculation of NMR shielding and indirect spin-spin coupling constants[J]. Chem Rev,1999,99(1):293-352.
[8] BELELLI P G,DAMIANI D E,CASTELLANI N J. DFT theoretical studies of UV-Vis spectra and solvent effects in olefin polymeriza- tion catalysts [J]. Chem Phys Lett,2005(401):515-521.
[9] CIURLA H,MICHALSKI J,HANUZA J,et al. Molecular structure,IR and Raman spectra as well as DFT chemical calculations for alkylaminoacetylureas:Vibrational characteristics of dicarbonylimide bridge[J]. Spectrochimica Acta Part A,2006,64(1):34- 46.
[10]徐璐.量化計算結合固體NMR研究高分子微觀結構、氫鍵與鏈運動[D].天津:南開大學,2010. XU L. Combined solid-state NMR and quantum chemical calculation studies of microstructure,hydrogen bond and segmental motion in polymers[D]. Tianjin:Nankai University,2010(in Chinese).
[11] FU W G,LI C,BAO H L,et al. Quantum chemical calculation on1H NMR chemical shifts of PMMA/PVPh blends[J]. Advanced Materials Research,2011(301/302/303):263-268.
[12]蘇永超,鄭安民,李申慧,等.藥物小分子化學位移的量子化學計算研究[J] .波譜學雜志,2006,23:294-301. SU Y C,ZHENG A M,LI S H,et al. Evaluation of quantum chemical methods for the calculation of13C NMR shifts of small drug molecules [J]. Chinese Journal of Magnetic Resonance,2006,23:294-301(in Chinese)
[13] DANG Q Q,LU S D,YU S,et al. Silk fibroin/montmorillonite nanocomposites:Effect of pH on the conformational transition and Clay dispersion[J]. Biomacromolecules,2010,11 (7):1796-1801.
[14]張榮純.發展固體核磁共振新方法及研究高分子復雜凝聚態結構與動力學[D].天津:南開大學,2014. ZHANG R C. Development of novel solid-state NMR methodsto study on complex condensed structures and dynamics of polymers[D]. Tianjin:Nankai University,2014(in Chinese).
[15] ZENG F,TONG Z,FENG H Q. NMR investigation of phase separation in poly(N-isopropyl acrylamide)/water solutions[J]. Polymer,1997,38(24):5539-5544.
Quantum chemical calculation on chemical shift of various carbons in typical polymer
FU Wei-gui,PAN Jing,LIU Shan-shan,FEI Mu-rong,CHEN Li
(State Key Laboratory of Separation Membranes and Membrane Processes,Tianjin Polytechnic University,Tianjin 300387,China)
Abstract:13C NMR chemical shifts of various carbons in polyphenylene oxide(PPO)and poly(N-isopropyl acylamine)(PNIPA)were calculated by Gaussian09 software based on density functional theory(DFT)with B3LYP//GIAO method at 6-311G(2d,p)level. Compared with the experimental results,the theoretical results accurately assigned the13C NMR chemical shifts of the carbons under different chemical environments. The results indicated that the chemical environments around the chemical groups changed with varying of molecular structures and its conformations.
Key words:quantum chemical calculation;NMR;chemical shift;density functional theory(DFT)
通信作者:付維貴(1980—),女,博士,副教授,主要研究方向為聚合物結構與性能. E-mail:tjfwy@hotmail.com
基金項目:國家自然科學基金資助項目(51303132);中國博士后基金項目(2014M551027);2015年度天津工業大學“研究生科技創新活動計劃”資助項目(15110)
收稿日期:2015-9-11
DOI:10.3969/j.issn.1671-024x.2016.02.011
中圖分類號:O641.3;O655
文獻標志碼:A
文章編號:1671-024X(2016)02-0056-04
