泮托拉唑鈉腸溶膠囊的質量控制及穩定性研究
2016-05-23 02:54:55尹美艷
安徽醫藥 2016年4期
尹美艷,高 蓉
(杭州中美華東制藥有限公司,浙江 杭州 310000)
?
泮托拉唑鈉腸溶膠囊的質量控制及穩定性研究
尹美艷,高蓉
(杭州中美華東制藥有限公司,浙江 杭州310000)
摘要:目的建立泮托拉唑鈉腸溶膠囊含量及有關物質的質量控制方法,并對上市品進行穩定性研究。方法以Agilent C(18)柱(250 nm×4.6 nm,5 μm)為色譜柱,流動相為磷酸二氫鈉緩沖(1.20 g的磷酸二氫鈉溶解于1 000 mL的水中,用磷酸調節pH至7.0)—乙腈(68∶32),流速1 mL·min(-1),檢測波長288 nm,對膠囊進行有關物質及含量的檢測;采用加速試驗,在溫度(40±2)℃、相對濕度(75±5)%的條件下放置6個月,考察其穩定性。結果泮托拉唑鈉在(1.0~10.0)×10(-3 ) g·L(-1)濃度范圍內線性關系良好,檢 測限為0.20 ng,定量限為0.60 ng,平均加樣回收率為100.57%(RSD=0.41%),測定樣品平均含量為100.67%,有關物質均小于0.2%。穩定性試驗結果含量在99.70%~101.16%,有關物質小于0.2% 。結論該方法快速,簡單,穩定,重現性好,可作為泮托拉唑鈉腸溶膠囊的含量及有關物質的檢測方法。穩定性檢測結果符合要求。
關鍵詞:泮托拉唑鈉腸溶膠囊;HPLC法;含量;有關物質;穩定性
泮托拉唑鈉是繼奧美拉唑鈉、蘭索拉唑鈉之后的為第三代質子泵抑制劑,由德國百克頓公司研制生產,于1994年在南非首次上市。化學名為:5-二氟甲氧基-2-[(3,4-二甲氧基-2-吡啶基)-甲基]亞硫酰基-1H-苯并咪唑鈉鹽一水合物,分子式為C16H12F2N3NaO4S·H2O。臨床主要用于治療急性消化性潰瘍(胃、十二指腸潰瘍)、反流性食管炎及卓—艾氏綜合征。 通過作用于H+/K+-ATP酶抑制胃酸和組胺分泌、作用時間長、起效快、強度大等優點成為治療潰瘍的一線藥物。抗潰瘍療效方面要明顯優于雷尼替丁,生物利用度高于奧美拉唑和蘭索拉唑[1-2]。泮托拉唑鈉腸溶膠囊國外藥典未收載,進口注冊標準及2015版《中國藥典》[3-4]收載了泮托拉唑鈉及泮托拉唑鈉腸溶膠囊的HPLC的檢驗方法,陳玉玲等雖報道過有關泮托拉唑鈉腸溶膠囊有關物質的檢測方法[5-7],通過試驗發現上述方法存在分離度低,重現性差等弊端,本文通過對HPLC條件的優化得到重現性好、分離度高、方便快捷的高效液相條件,使得泮托拉唑鈉腸溶膠囊質量得到有效控制。
1儀器與試藥
Agilent 1260型高效液相色譜儀(DAD),安捷倫色譜工作站,電子分析天平(XSE 105DU)。色譜純乙腈(Fisher公司),重蒸餾水,磷酸二氫鈉(分析純,國藥集團化學試劑有限公司),磷酸(分析純,國藥集團化學試劑有限公司),氫氧化鈉(分析純,國藥集團化學試劑有限公司)。泮托拉唑鈉對照品(中國食品藥品檢定研究院,批號100575-201408),泮托拉唑鈉腸溶膠囊(山東綠葉制藥有限公司,批號14110202,15021001,15031802,規格40 mg)。
2方法和結果
2.1色譜條件色譜柱:Agilent C18柱(250 nm×4.6 nm,5 μm);流動相:磷酸二氫鈉緩沖(1.20g的磷酸二氫鈉溶解于1 000 mL的水中,用磷酸調節pH至7.0)-乙腈(68∶32);流速:1 mL·min-1;檢測波長:288 nm;進樣量:20 μL;柱溫:40℃。
2.2溶液配制
2.2.1含量測定對照品溶液取泮托拉唑鈉對照品適量,加入0.001 mol·L-1氫氧化鈉溶液-乙腈(1∶1)混合溶劑溶解并定量稀釋至每1 mL含有0.04 mg的溶液,備用。
2.2.2含量測定供試品溶液取泮托拉唑鈉腸溶膠囊20粒,取內容物研細,精密稱量適量(約相當于泮托拉唑鈉40 mg),置100 mL量瓶中,加入0.001 mol·L-1氫氧化鈉溶液—乙腈(1∶1)混合溶劑,超聲使泮托拉唑鈉溶解,補加上述溶劑稀釋至刻度,搖勻,取濾液10 mL,置100 mL量瓶中,加上述溶劑稀釋至刻度,搖勻,作為供試品溶液備用。
2.2.3 有關物質測定供試品溶液稱取“2.2.2”項中泮托拉唑鈉腸溶膠囊內容物適量,研細,取粉末精密稱定,置50 mL的量瓶中,加入0.001 mol·L-1氫氧化鈉溶液—乙腈(1∶1)混合溶劑適量,超聲溶解,加入上述溶液稀釋至刻度,搖勻。配制成1 mL中含有約0.4 mg泮托拉唑鈉的溶液。
2.2.4 有關物質對照溶液精密量取“2.2.3”項中的有關物質供試品溶液1 mL,置200 mL的量瓶中,加入0.001 mol·L-1氫氧化鈉溶液—乙腈(1∶1)混合溶劑稀釋至刻度,搖勻。
2.2.5 陰性對照溶液按照處方比例稱取羥丙甲纖維素(Hydroxypropyl Methyl Cellulose,HPMC )、吐溫-80(Tween-80)相當于一粒膠囊的輔料量,置于100 mL量瓶中,加0.001 mol·L-1氫氧化鈉溶液—乙腈(1∶1)混合溶劑稀釋至刻度,超聲,濾過,取1 mL加入上述溶劑稀釋與樣品相同倍數,即得。
2.3方法學考察
2.3.1專屬性試驗取“2.2.1”項下泮托拉唑鈉對照品溶液精密量取 20 μL進色譜儀,按照上述色譜條件測定,記錄色譜圖。按照上述方法分別取“2.2.2”項下含量供試品溶液、 “2.2.3”項有關物質供試品溶液、“2.2.4”項有關物質對照及“2.2.5”陰性對照品溶液,稀釋至同等濃度,進色譜儀測定,記錄色譜圖,結果輔料峰未干擾測定(圖1)。分別取泮托拉唑鈉腸溶膠囊適量,取內容物研細,精密稱定粉末(約相當于主藥40 mg)進行如下測試:(1)酸破壞:將精密稱定后的粉末置具塞試管中,用0.1 mol·L-1鹽酸溶液25 mL,搖勻,置于100℃水浴鍋中4 h,放冷。加入0.1 mol·L-1氫氧化鈉中和并轉移至100 mL的容量瓶中,用0.001 mol·L-1氫氧化鈉溶液—乙腈(1∶1)混合溶劑稀釋至刻度,搖勻,過濾。(2)堿破壞:將精密稱定后的粉末置具塞試管中,用0.1 mol·L-1氫氧化鈉溶液25 mL,搖勻,置于100℃水浴鍋中4 h,放冷。加入0.1 mol·L-1鹽酸中和并轉移至100 mL的容量瓶中,用0.001 mol·L-1氫氧化鈉溶液—乙腈(1∶1)混合溶劑稀釋至刻度,搖勻,過濾。(3)氧化破壞:將精密稱定后的粉末置具塞試管中,加入30%過氧化氫溶液10 mL,搖勻,放置12 h,加0.001 mol·L-1氫氧化鈉溶液—乙腈(1∶1)混合溶劑稀釋至刻度,搖勻,過濾。(4)加熱破壞:將精密稱定后的粉末置具塞試管中,加0.001 mol·L-1氫氧化鈉溶液-乙腈(1∶1)混合溶劑稀釋,配置成濃度0.04 g·L-1溶液25 mL,置于100℃水浴鍋中4 h,放冷,加上述溶液轉移至100 mL容量瓶中,用上述溶液稀釋至刻度,搖勻,過濾。(5)光破壞:將精密稱定后的粉末置具塞試管中,加0.001 mol·L-1氫氧化鈉溶液-乙腈(1∶1)混合溶劑稀釋,配置成0.04 g·L-1溶液25 mL,置于強光(5 000 Lx)下照射12 h后,放冷。按照上述色譜條件進行測定,記錄色譜圖。結果顯示樣品在經過酸、堿、氧化、加熱、光照破壞后主成分與各降解產物可以有效分離(圖2)。

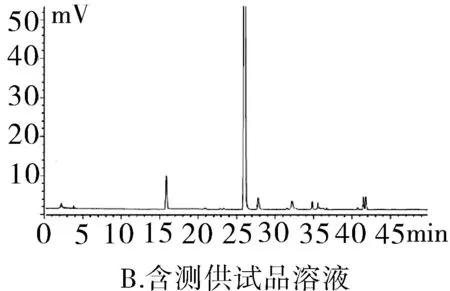
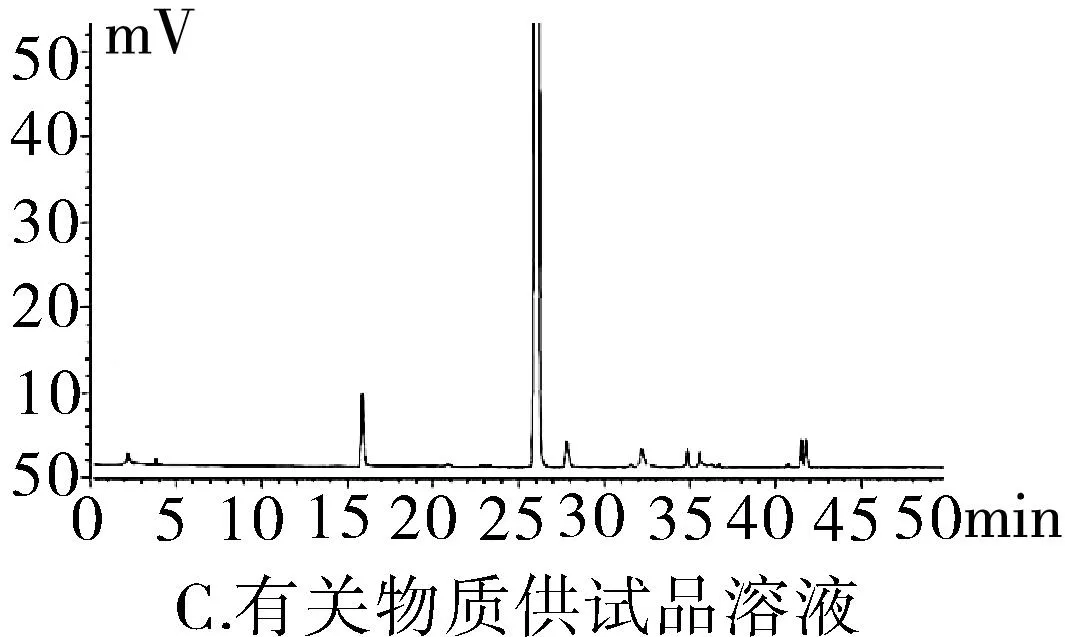
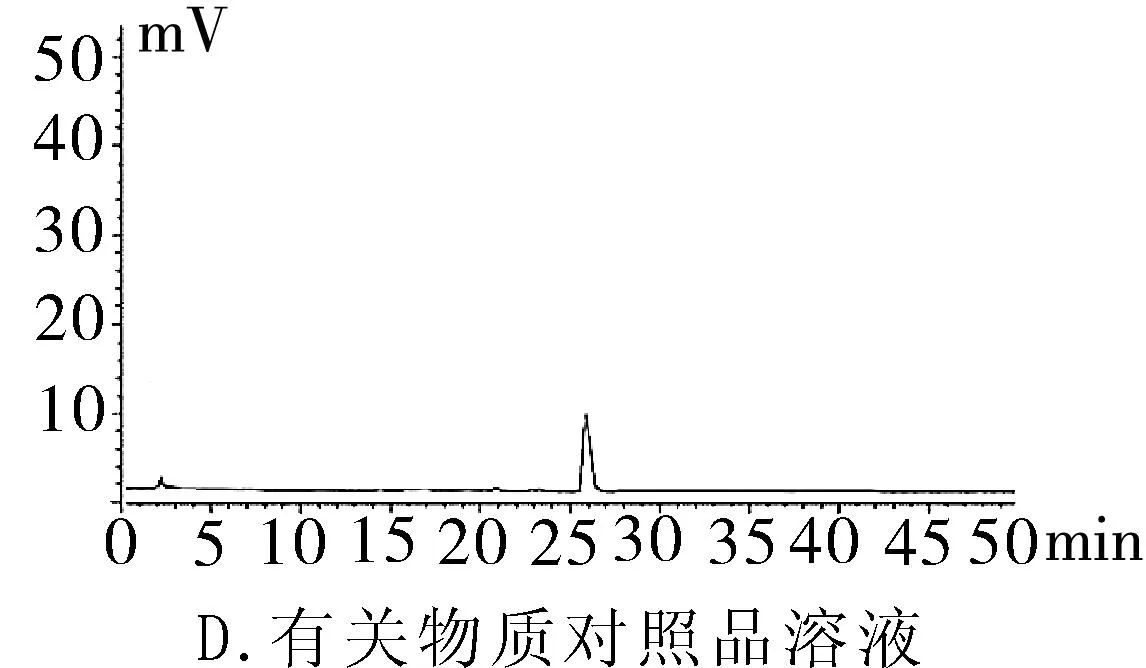

圖1 HPLC圖譜
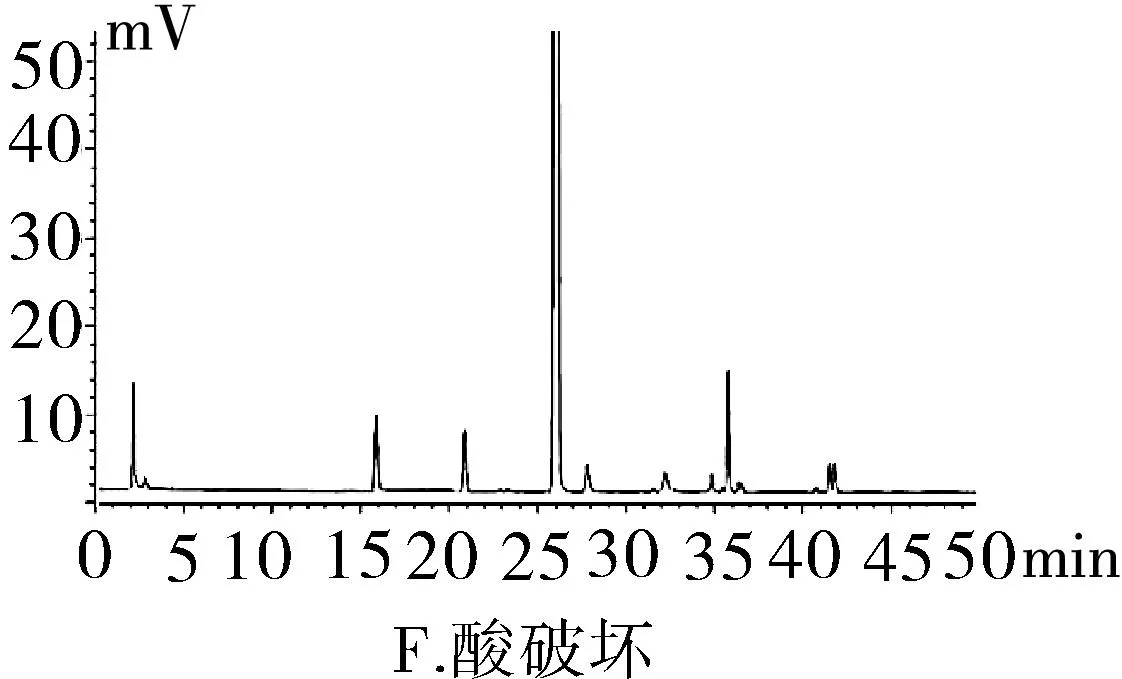
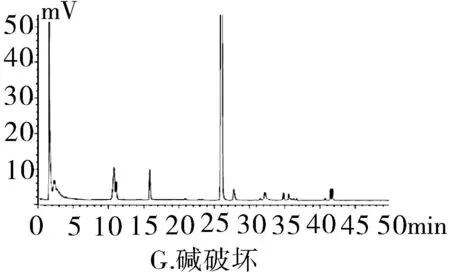



圖2 泮托拉唑鈉方法學圖譜
2.3.2系統適用性試驗分別精密量取“2.2.1”項中泮托拉唑鈉對照品溶液、“2.2.2”泮托拉唑鈉腸溶膠囊供試品溶液及“2.2.5”項陰性對照溶液20 μL,按上述色譜條件進樣測定,色譜柱的理論塔板數按照泮托拉唑鈉計算不得少于3 000,泮托拉唑峰與相鄰雜質峰分離度不低于2.0。
2.3.3檢測限及定量限測定取“2.2.1”項中配制的泮托拉唑鈉對照品溶液,稀釋至0.4×10-3g·L-1,按照上述色譜條件進樣,根據信噪比法進行試驗,得到的結果:檢測限為0.20 ng(S/N=3),定量限為0.60 ng(S/N=10)。
2.3.4精密度及重復性試驗取“2.2.1”項下泮托拉唑鈉對照品溶液,按照上述色譜條件進樣,重復測定6次,記錄泮托拉唑鈉的峰面積,計算相對標準偏差RSD=0.31%,表明儀器精密度良好。

表1 加樣回收率試驗
取14110202批次泮托拉唑鈉腸溶膠囊6粒,分別取內容物研細,精密稱量適量粉末(相當于主藥20 mg),加0.001 mol·L-1氫氧化鈉溶液—乙腈(1∶1)混合溶劑配制成濃度為0.4 g·L-1供試品溶液6份,按照上述色譜條件在同一高效液相色譜儀上分別進樣,記錄泮托拉唑鈉峰面積,計算相對標準偏差RSD=0.37%,顯示重現性良好。
2.3.5加樣回收率試驗按照處方比例稱量適量輔料(相當于1粒膠囊輔料的量),分別加入處方量的80%,100%,120%的泮托拉唑鈉的對照品,加0.001 mol·L-1氫氧化鈉溶液—乙腈(1∶1)混合溶劑溶解并稀釋至刻度,搖勻,每個含量的溶液分別取3份作為供試品溶液。精密量取20 μL溶液,按照上述色譜條件進高效液相色譜儀,記錄圖譜,結果見表1。計算得平均加樣回收率為100.57%,RSD=0.41%。
2.3.6樣品溶液穩定性試驗取14110202批次樣品制備成濃度為0.4 g·L-1供試品溶液,分別在0、2、4、6、12、18 h精密吸取20 μL進樣,記錄色譜圖,根據泮托拉唑鈉峰面積,計算其相對標準偏差RSD為0.07%,說明在18 h內泮托拉唑鈉在溶液內穩定。
2.4樣品測定
2.4.1有關物質測定取3批上市品泮托拉唑鈉腸溶膠囊各10粒,取內容物研細,精密稱量適量粉末(相當于泮托拉唑鈉40 mg),置100 mL的量瓶中,加0.001 mol·L-1氫氧化鈉溶液—乙腈(1∶1)混合溶劑振蕩溶解,并稀釋至刻度,搖勻,過濾,濾液作為供試品。取“2.2.1”項中泮托拉唑鈉對照品溶液稀釋與供試品同等濃度作為對照溶液。精密量取供試品和對照品溶液20 μL進高效液相色譜儀,記錄色譜圖。根據面積歸一化法計算有關主峰及有關物質的峰面積。
2.4.2含量測定取3批次上市品泮托拉唑鈉腸溶膠囊各10粒,取內容物研細,制備成濃度同2.2.1項中濃度作為供試品溶液。分別精密量取“2.2.1”項中泮托拉唑鈉對照品溶液稀釋與供試品各20 μL,進高效液相色譜儀,記錄圖譜。按照外標法分別測定測定3批上市品含量。3批樣品的有關物質及含量的結果見表2。
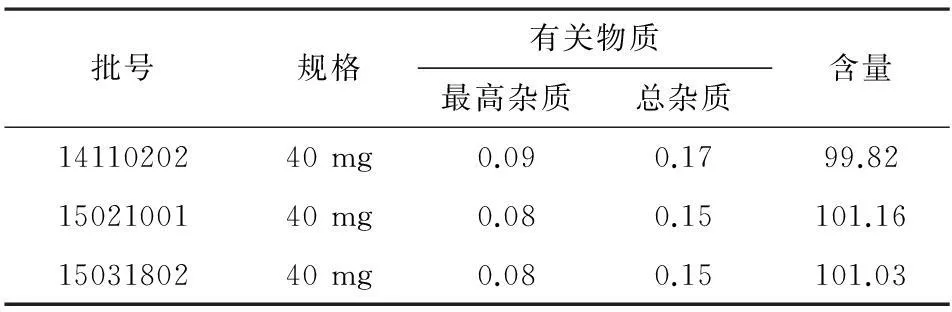
表2 泮托拉唑鈉腸溶膠囊有關物質及含量測定結果

表3 泮托拉唑鈉腸溶膠囊的穩定性果
2.5穩定性研究參照《中國藥典》2015版附錄9001 原料藥物與制劑穩定性試驗指導原則,對市售的3批制劑(14110202,15021001,15031802)進行加速試驗,取市售最小包裝,在溫度(40±2)℃、相對濕度(75±5)%的條件下放置6個月。分別于1月、2月、3月和6月取樣一次,按照“2.4”項中有關物質及含量的測定方法對泮托拉唑鈉腸溶膠囊的穩定性進行考察,結果見表3。
3討論
泮托拉唑鈉因含有亞磺酰基苯并咪唑機構,導致產品對熱、光不穩定,在酸性條件下不穩定[8],在做產品的方法學時,選擇0.001 mol·L-1氫氧化鈉溶液—乙腈(1∶1)的混合溶液溶解測定,避免在偏酸或中性的溶劑中產生降解雜質。
本文通過對比《中國藥典》提供的Kromasil、Waters及Agilent的C18柱,通過在同一儀器上的驗證得到Agilent的C18柱具有分離度高、柱效穩定,能有效分離雜質優點,并且在288 nm,主峰和破壞雜質峰有最大吸收。通過對泮托拉唑鈉腸溶膠囊的測定結果,在擬定的HPLC條件下,主峰、輔料峰及有關物質能夠達到基線分離,符合檢測要求。方便簡便,快捷,且能滿足質量控制要求。
通過對3批不同批次的市售藥品進行了6個月加速試驗,從測定結果可以看出,產品穩定性較好,在制劑的條件下能有效避免原料藥不穩定的因素,說明產品的制劑工藝成熟、穩定,能有效控制雜質,保證含量,對藥物在臨床的使用提供安全保障。
參考文獻:
[1]郁心圃. 泮托拉唑鈉和奧美拉唑治療胃潰瘍的對照研究[J].實用臨床醫藥雜志,2012,16(19):97-98.
[2]張俊平.奧美拉唑、蘭索拉唑、泮托拉唑治療胃潰瘍臨床療效觀察[J].現代診斷與治療,2015,26(4):800-801.
[3]國家食品藥品監督管理局.進口藥品注冊標準JX20070162[S].
[4]國家藥典委員會.中國藥典(二部)[S].北京:中國醫藥科技出版社,2015:705.
[5]陳玉玲.HPLC法測定泮托拉唑鈉腸溶微丸膠囊的含量[J].科技創新與應用,2012(1):38.
[6]馬力,黃良升,張玉.高效液相色譜法測定泮托拉唑鈉腸溶微丸膠囊的含量[J].醫學導報,2009,28(7):924-925.
[7]張舒,張昀.HPLC梯度洗脫法測定泮托拉唑鈉腸溶膠囊的有關物質[J].中國藥師,2015,18(6):943-945.
[8]王婧思,李桂龍,王成港,等.泮托拉唑鈉有關物質分析方法的比較[J].藥物評價研究,2012,35(2):109-112.
The study of quality and stability on pantoprazole sodium enteric-coated capsules
YIN Mei-yan,GAO Rong
(HuadongMedicineCo.,LTD.,Hangzhou,Zhejiang310000)
Abstract:ObjectiveAn HPLC method was established for determination of the content of Pantoprazole Sodium Enteric-Coated Capsules and Its Related Substances,and study the stability .Methods The chromatographic separation was performed on AgilentC(18)(250 nm×4.6 nm,5 μm),Sodium dihydrogen phosphate buffer(dissolved 1.20 g sodium dihydrogen phosphate in 1 000 mL water,and ajust pH 7.0 with phosphoric acid)—acetonitrile=68∶32,the flow rate was 1.0 mL·min(-1),the detection wavelength was 288 nm,and detective the related substances and the content . Theacceleration test was carried outat temperature (40±2)℃,relative humidity of (75±5)%,and test the result.ResultsThe linear of Pantoprazole Sodium of atorvastatin calcium was (1.0~10.0)×10(-3) g·L(-1),the limits of detection was 0.20 ng,the limit of quantitation was 0.60 ng,the average recovery was 100.57%(RSD=0.41%),Determination of the sample average content was 100.67%,and the related substance was less than 0.2%. The result of stability test that the content was 99.70%~101.16%,the related substance was less than 0.2%.ConeclusionThe method was quick,simple,stable,good repeatability,and it could be used as the content of pantoprazole sodium enteric-coated capsules and its related substances. The stability results conform to the requirement.
Key words:pantoprazole sodium enteric-coated capsules;HPLC;content;related substances;stability
(收稿日期:2015-10-16,修回日期:2015-12-21)
doi:10.3969/j.issn.1009-6469.2016.04.013
