ATP敏感性鉀通道在硫化氫抑制壞死性凋亡介導的高糖致H9c2心肌細胞炎癥中的作用*
2016-06-01 11:31:17梁偉杰陳美姬何健華張穩柱馮鑒強黃惠敏
中國病理生理雜志 2016年8期
梁偉杰, 陳美姬, 何健華, 張穩柱, 程 飛, 蘭 軍, 馮鑒強, 黃惠敏△
(1廣州市番禺區中心醫院心血管內科,2廣州市番禺區心血管疾病研究所,廣東 廣州 511400;3中山大學附屬第一醫院黃埔院區兒科,廣東 廣州 510700; 4廣州市番禺區石碁人民醫院心血管內科,廣東 廣州 511450; 5東莞市第三人民醫院心血管內科,6東莞市心血管疾病研究所,廣東 東莞 523326)
ATP敏感性鉀通道在硫化氫抑制壞死性凋亡介導的高糖致H9c2心肌細胞炎癥中的作用*
梁偉杰1, 2, 陳美姬3, 何健華4, 張穩柱1, 2, 程 飛5, 6, 蘭 軍5, 6, 馮鑒強5, 6, 黃惠敏1, 2△
(1廣州市番禺區中心醫院心血管內科,2廣州市番禺區心血管疾病研究所,廣東 廣州 511400;3中山大學附屬第一醫院黃埔院區兒科,廣東 廣州 510700;4廣州市番禺區石碁人民醫院心血管內科,廣東 廣州 511450;5東莞市第三人民醫院心血管內科,6東莞市心血管疾病研究所,廣東 東莞 523326)
目的: 探討ATP敏感性鉀通道(KATP通道)在硫化氫(H2S)抑制壞死性凋亡介導的高糖(HG)致H9c2 心肌細胞炎癥中的作用。方法: 應用Western blot法測定受體相互作用蛋白3(RIP3)和環氧化酶-2(COX-2)的表達水平;ELISA檢測細胞培養液中白細胞介素-1β(IL-1β)和腫瘤壞死因子-α (TNF-α)的水平。結果: H9c2 心肌細胞經HG (35 mmol/L葡萄糖)處理24 h,其RIP3 的表達水平明顯升高,100 μmol/L KATP通道開放劑二氮嗪(DZ)和400 μmol/L H2S的供體硫氫化鈉(NaHS)預處理心肌細胞30 min均可抑制HG對RIP3 表達的上調;100 μmol/L KATP通道阻斷劑5-羥基癸酸(5-HD)預處理心肌細胞30 min可阻斷NaHS對HG上調RIP3 表達的抑制作用。另一方面,100 μmol/L壞死性凋亡的特異性抑制劑necrostatin-1共處理或100 μmol/L DZ、400 μmol/L NaHS預處理心肌細胞均能抑制高糖引起的心肌細胞炎癥,使COX-2 表達及IL-1β和TNF-α的分泌水平均減少;而100 μmol/L 5-HD能明顯拮抗NaHS的上述抗炎癥反應作用。結論: KATP通道在H2S抑制壞死性凋亡介導的高糖致心肌細胞炎癥反應中發揮重要的作用。
ATP敏感性鉀通道; 硫化氫; 壞死性凋亡; 高糖; 心肌細胞; 炎癥
糖尿病心肌病(diabetic cardiomyopathy,DCM)是一種嚴重的糖尿病心血管并發癥,是引起糖尿病病人心力衰竭和死亡的主要原因,受到全球的廣泛關注。DCM的發病涉及多方面的機制,如氧化應激、心肌纖維化、慢性炎癥和心肌細胞死亡等[1]。研究表明,在DCM患者或動物模型中,持續存在的炎癥反應伴隨大量的炎癥細胞因子如白細胞介素(interleukin,IL)-1β、IL-6、腫瘤壞死因子-α(tumor necrosis factor-α,TNF-α)的分泌及與炎癥相關的信號分子,如核因子-κB(nuclear factor κB,NF-κB)、環氧化酶-2(cyclooxygenase-2,COX-2)等的激活,最終導致心肌損傷[2-4]。另一方面,心肌細胞死亡在DCM的發生過程中起關鍵的開啟作用。壞死性凋亡(necroptosis)也稱為程序性壞死(programmed necrosis),是近年來新發現的一種細胞死亡方式。在壞死性凋亡發生過程中,受體相互作用蛋白(receptor-interacting protein,RIP)家族,尤其是RIP3起重要的作用,過量表達的RIP3可導致壞死性凋亡的發生[5]。有報道指出,壞死性凋亡和細胞炎癥反應的關系非常密切。Liu等[6]證實,壞死性凋亡的特異性抑制劑necrostatin-1(Nec-1)可減輕小鼠結腸癌細胞的炎癥反應,提示壞死性凋亡可介導細胞的炎癥反應。但是,在DCM的病理生理機制中,壞死性凋亡和炎癥的關系如何目前尚未完全明確,因此,研究這個問題有重要的理論和臨床意義。
ATP敏感性鉀通道(ATP-sensitive K+channel,KATP通道)是Noma于1983年首先在豚鼠的心肌細胞中發現的,廣泛存在于心肌細胞、骨骼肌細胞和血管平滑肌細胞中。多項研究表明,開放KATP通道對心肌有明確的保護作用[7-9]。新型氣體信號分子硫化氫(hydrogen sulfide,H2S)是KATP通道的開放劑[10],能保護H9c2心肌細胞對抗高糖引起的心肌細胞毒性、凋亡、氧化應激、線粒體功能受損及炎癥等損傷[11-14]。最近,我們證實,H2S通過調控KATP通道[12]及抑制壞死性凋亡對抗高糖引起的心肌細胞損傷[14],然而,KATP通道是否參與H2S對炎癥反應及壞死性凋亡的抑制,迄今未見報道。
為此,本研究建立高糖損傷H9c2心肌細胞模型[11],重點探討以下問題:(1)壞死性凋亡是否介導高糖致心肌細胞炎癥反應;(2)開放KATP通道能否抑制壞死性凋亡介導的炎癥反應;(3)KATP通道在H2S抑制壞死性凋亡介導的心肌細胞炎癥反應中的作用。
材 料 和 方 法
1 材料
Nec-1和硫氫化鈉(sodium hydrosulfide,NaHS)由Sigma-Aldrich供應;抗RIP3 抗體和抗COX-2 抗體購自CST;二氮嗪(diazoxide, DZ)和5-羥基癸酸(5-hydroxydecanoic acid,5-HD)購自Cayman;特級胎牛血清(fetal bovine serum,FBS)購自Gibco BRL;DMEM培養基(其葡萄糖濃度為5.5 mmol/L)由HyClone供應;TNF-α和IL-1β ELISA試劑盒由武漢華美生物工程有限公司提供。H9c2心肌細胞來源于胚胎期大鼠心臟組織的亞克隆細胞系,由中山大學實驗動物中心供應。
2 方法
2.1 細胞培養 H9c2心肌細胞置于含10% FBS的DMEM培養基中,于5% CO2、37 ℃的條件下傳代培養,待細胞生長至約80%的融合狀態可用于實驗。
2.2 實驗分組 實驗分為10組:(1)正常對照(control)組采用DMEM培養基作用心肌細胞24 h;(2)高糖(high glucose,HG)組用含有高濃度葡萄糖(35 mmol/L)的DMEM培養基作用心肌細胞24 h;(3) NaHS+HG組用400 μmol/L NaHS預處理心肌細胞30 min,PBS液洗2次后,用含高濃度葡萄糖的DMEM培養基作用24 h;(4) 5-HD+NaHS+HG組用100 μmol/L 5-HD作用心肌細胞30 min,撤去,PBS洗2次,后續步驟與第(3)實驗組相同;(5) DZ+HG組用100 μmol/L DZ預處理心肌細胞30 min,PBS液洗2次后,用含高濃度葡萄糖的DMEM培養基作用24 h;(6) Nec-1+ HG組用100 μmol/L Nec-1與含高濃度葡萄糖的DMEM培養基共處理心肌細胞24 h;(7) NaHS組用400 μmol/L NaHS作用心肌細胞30 min,PBS洗2次,接著DMEM培養基處理24 h;(8) 5-HD組用100 μmol/L 5-HD作用心肌細胞30 min,PBS洗2次,接著DMEM培養基處理24 h;(9) DZ組用100 μmol/L DZ作用心肌細胞30 min,撤去,PBS洗2次,接著DMEM培養基處理24 h;(10) Nec-1組用100 μmol/L Nec-1與DMEM培養基共處理心肌細胞24 h。
2.3 Western blot法檢測RIP3 和COX-2 的表達水平 在60 mm培養皿中種植H9c2 心肌細胞,待細胞生長至大約80%的融合度時,根據實驗分組給予相應處理后,加入細胞裂解液,4 ℃的搖床上作用30 min,12 000 r/min高速離心10 min,以BCA法測定蛋白質含量。等量的蛋白經10%SDS-PAGE分離后,轉移至PVDF膜上,5%脫脂奶粉封閉1 h,加入Ⅰ抗,即兔抗鼠RIP3、COX-2或GAPDH(濃度均為1∶1 000),4 ℃作用過夜后加入濃度為1∶2 500的Ⅱ抗稀釋液,室溫下孵育1.5 h,ECL法使PVDF膜顯色,暗室中將顯色條帶曝光到醫用X線片上,凝膠成像掃描系統分析結果。實驗重復5次。
2.4 ELISA檢測細胞培養液中IL-1β和TNF-α的水平 于96孔板中種植H9c2 心肌細胞,待細胞融合度達到約80%時,按照分組給予不同的處理后,收集培養基留作待測標本。ELISA操作流程根據試劑盒說明書進行,在終止顯色反應后,用酶標儀測定各孔450 nm處吸光度(A)值。取5孔A值的平均數,根據以下公式計算出IL-1β和TNF-α的誘導釋放率(%):處理組A/對照組A×100%。實驗重復5次。
3 統計學處理
應用SPSS 17.0軟件進行統計數據處理和分析,計量資料采用均數±標準誤(mean±SEM)表示,單因素方差分析(one-way ANOVA)用于多個樣本均數間的比較,SNK-q檢驗用于多個樣本均數間的兩兩比較,以P<0.05為差異有統計學意義。
結 果
1 KATP通道開放劑抑制HG誘導的心肌細胞RIP3表達增加
圖1顯示,H9c2心肌細胞經HG (35 mmol/L葡萄糖)作用24 h后,其RIP3的表達明顯增加,與control組比較,差異有統計學顯著性(P<0.01)。應用100 μmol/L DZ(KATP通道開放劑)預處理心肌細胞30 min后再予HG作用心肌細胞24 h,RIP3的表達明顯減少,與HG組比較,差異有統計學顯著性(P<0.01)。100 μmol/L DZ本身對心肌細胞RIP3的基礎表達無明顯影響。

Figure 1.KATPchannel opener diazoxide (DZ) attenuated high glucose (HG)-induced up-regulation of RIP3 in H9c2 cardiac cells. Mean±SEM.n=5.**P<0.01vscontrol group;##P<0.01vsHG group.
圖1 KATP通道開放劑抑制HG誘導的心肌細胞RIP3表達增加
2 KATP通道介導H2S對HG上調RIP3表達的抑制作用
如前所述,HG可明顯上調RIP3的表達;在HG處理心肌細胞前,應用400 μmol/L NaHS預處理心肌細胞30 min可使RIP3的表達明顯減少,與HG處理組比較,差異有統計學顯著性(P<0.01)。但是,在NaHS預處理前,應用100 μmol/L 5-HD(KATP通道阻斷劑)作用心肌細胞30 min,可使NaHS對RIP3表達的抑制作用減弱,與NaHS預處理組比較,差異有統計學顯著性(P<0.01)。400 μmol/L NaHS或100 μmol/L 5-HD本身對心肌細胞RIP3的基礎表達無明顯的影響,見圖2。
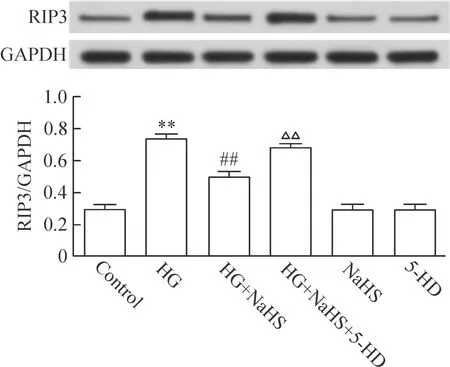
Figure 2.KATPchannels contributed to the inhibitory effect of H2S on HG-induced increase in RIP3 expression in H9c2 cardiac cells. Mean±SEM.n=5.**P<0.01vscontrol group;##P<0.01vsHG group;△△P<0.01vsHG+NaHS group.
圖2 KATP通道介導H2S抑制HG誘導的心肌細胞RIP3表達增加
3 壞死性凋亡抑制劑和KATP通道開放劑抑制HG誘導的心肌細胞COX-2 表達上調
HG作用心肌細胞24 h可明顯上調COX-2的表達,與對照組比較,兩者差異有統計學顯著性(P<0.01)。但是,應用100 μmol/L Nec-1(壞死性凋亡的特異性抑制劑)和HG共處理心肌細胞24 h可明顯拮抗HG對COX-2表達的上調作用,與HG組比較,差異有統計學顯著性(P<0.01)。100 μmol/L Nec-1本身對心肌細胞COX-2的基礎表達無明顯影響。
與Nec-1的作用相類似,100 μmol/L DZ預處理心肌細胞也能明顯地抑制HG對COX-2表達的上調,與HG組比較,差異有統計學顯著性(P<0.01)。100 μmol/L DZ本身對心肌細胞COX-2的基礎表達無明顯影響,見圖3。

Figure 3.The inhibitor of necroptosis (A) and KATPchannel opener (B) attenuated HG-induced up-regulation of COX-2 in H9c2 cardiac cells. Mean±SEM.n=5.**P<0.01vscontrol group;##P<0.01vsHG group.
圖3 壞死性凋亡抑制劑和KATP通道開放劑抑制HG誘導的心肌細胞COX-2表達上調
4 KATP通道介導H2S對HG促進COX-2表達的抑制作用
如前所述,HG可促進COX-2的表達;應用400 μmol/L NaHS預處理心肌細胞30 min,可使COX-2 的表達明顯減少,與HG處理組比較,差異有統計學顯著性(P<0.01)。但是,在NaHS預處理前,應用100 μmol/L 5-HD預處理心肌細胞30 min可使NaHS對COX-2表達的抑制作用明顯減弱,與NaHS預處理組比較,差異有統計學顯著性(P<0.01)。400 μmol/L NaHS或100 μmol/L 5-HD本身對心肌細胞COX-2的基礎表達無明顯的影響,見圖4。

Figure 4.KATPchannels contributed to the inhibitory effect of H2S on HG-induced increase in the expression of COX-2 in H9c2 cardiac cells. Mean±SEM.n=5.**P<0.01vscontrol group;##P<0.01vsHG group;△△P<0.01vsHG+NaHS group.
圖4 KATP通道介導H2S抑制HG誘導的心肌細胞COX-2 表達上調
5 KATP通道參與H2S對壞死性凋亡介導的HG致炎癥細胞因子分泌的抑制
如圖5所示,HG處理心肌細胞24 h可明顯促進炎癥細胞因子IL-1β和TNF-α的分泌,與control組比較,差異均有統計學顯著性(P<0.01)。應用100 μmol/L Nec-1共處理或100 μmol/L DZ預處理心肌細胞能拮抗HG對炎癥細胞因子分泌的促進作用,使IL-1β和TNF-α的分泌水平降低,與HG組分別比較,差異均有統計學顯著性(P<0.01)。100 μmol/L Nec-1或100 μmol/L DZ本身不影響炎癥細胞因子的基礎分泌。
與Nec-1或DZ抑制炎癥細胞因子分泌的作用相類似,應用400 μmol/L NaHS預處理心肌細胞30 min也可使IL-1β和TNF-α的分泌水平明顯降低,與HG組比較,差異均有統計學顯著性(P<0.01)。但是,在NaHS預處理前,應用100 μmol/L 5-HD作用心肌細胞30 min可明顯拮抗NaHS對HG誘導炎癥細胞因子分泌的抑制作用,使IL-1β和TNF-α的分泌水平升高,與NaHS預處理組比較,差異均有統計學顯著性(P<0.01)。400 μmol/L NaHS或100 μmol/L 5-HD本身對炎癥細胞因子的基礎分泌無明顯的影響。
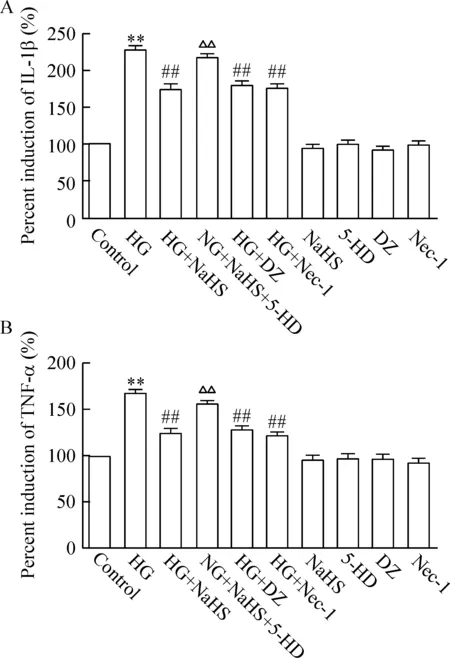
Figure 5.KATPchannels mediated the inhibitory effect of H2S on HG-induced secretion of inflammatory cytokines mediated by necroptosis in H9c2 cardiac cells. Mean±SEM.n=5.**P<0.01vscontrol group;##P<0.01vsHG group;△△P<0.01vsHG+NaHS group.
圖5 KATP通道參與H2S抑制壞死性凋亡介導的高糖致心肌細胞炎癥細胞因子分泌
討 論
炎癥反應是高血糖引起心肌損傷的一個重要的病理生理機制。和我們之前的報道[12]及其它的研究結果[2-4]相一致,本研究再次證實,HG可引起明顯的心肌細胞炎癥反應,表現為炎癥細胞因子IL-1β、TNF-α的分泌水平升高和炎癥通路COX-2的激活。生理狀態下COX-2在絕大部分組織細胞中是鮮有表達的,在炎癥、缺氧、高血糖、腫瘤等病理狀態下則表達呈增高趨勢,誘導合成前列腺素類衍生物,是誘導炎癥反應的一種重要的信號分子。研究表明,細胞炎癥和壞死性凋亡的關系非常密切。最近,我們證實壞死性凋亡參與HG引起的心肌細胞損傷[14]。但是,壞死性凋亡是否參與HG引起的心肌細胞炎癥尚不清楚。因此,在本研究中,我們觀察了壞死性凋亡的特異性抑制劑Nec-1對HG誘導的心肌細胞炎癥的影響,結果表明Nec-1可明顯抑制HG引起的心肌細胞炎癥反應,使炎癥細胞因子的分泌和COX-2的表達減少,清晰地提示壞死性凋亡可介導HG誘導的H9c2心肌細胞炎癥。Liu等[6]在結腸癌細胞(HT-29細胞)模型的研究結論與本文相似,且我們首次證實,在HG損傷心肌細胞的條件下,壞死性凋亡可激活COX-2通路,這可能是壞死性凋亡介導炎癥的一個重要機制,但尚需進一步的探討。
KATP通道是一種受細胞內ATP濃度調控的內向整流鉀通道,其活性狀態主要決定于胞內ATP與ADP的濃度。心肌細胞KATP通道的激活可通過縮短動作電位時程、減少鈣超載、減少能量消耗、抑制線粒體滲透性轉換孔開放等機制,發揮心肌保護作用。因壞死性凋亡可引起細胞內ATP濃度的變化[15],因此我們推測KATP通道可能對壞死性凋亡有一定的調控作用。為了驗證我們的假設,我們首先觀察了KATP通道開放劑DZ對HG上調RIP3表達的影響。結果表明,DZ可明顯地拮抗HG對RIP3 表達的上調作用。此外,應用DZ預處理心肌細胞和應用壞死性凋亡的特異性抑制劑Nec-1共處理心肌細胞均產生類似的抗炎癥反應作用,使IL-1β、TNF-α的分泌和COX-2的表達減少。上述結果提示,開放KATP通道可抑制壞死性凋亡及其介導的心肌細胞炎癥反應。
重要的是,我們進一步探討了高糖狀態下,KATP通道在H2S抑制壞死性凋亡介導的心肌細胞炎癥中的作用。H2S是繼CO和NO之后被發現的第3種內源性氣體信號分子,具有多種病理生理作用。H2S是KATP通道的開放劑[10],可通過調控KATP通道實現細胞保護作用。例如,H2S可激活KATP通道減輕缺氧對神經母細胞瘤細胞(SH-SY5Y細胞)的損傷[16];Bian等[17]報道,KATP通道介導H2S對缺血引起大鼠心肌細胞損傷的抑制作用;我們最近證實,H2S可通過開放KATP通道對抗HG引起的H9c2心肌細胞損傷[13]。為了探討KATP通道是否介導H2S對HG誘發的心肌細胞炎癥的抑制作用,本研究在上述研究的基礎上,進一步觀察KATP通道阻斷劑5-HD預處理心肌細胞對H2S抑制HG致心肌細胞炎癥反應的影響。結果表明,5-HD可明顯減弱H2S的抗壞死性凋亡和抗炎癥反應作用,使RIP3和COX-2表達水平明顯上升,IL-1β和TNF-α分泌增多,提示在高糖狀態下,通過激活KATP通道繼而抑制壞死性凋亡介導的心肌細胞炎癥反應,可能是H2S發揮其保護心肌細胞對抗HG引起的炎癥反應的重要機制之一,這為深入闡明H2S的抗炎癥機制提供了新穎的實驗依據。
[1] Falc?o-Pires I, Leite-Moreira AF. Diabetic cardiomyopathy: understanding the molecular and cellular basis to progress in diagnosis and treatment[J]. Heart Fail Rev, 2012, 17(3):325-344.
[2] Wen HL, Liang ZS, Zhang R, et al. Anti-inflammatory effects of triptolide improve left ventricular function in a rat model of diabetic cardiomyopathy[J]. Cardiovasc Diabetol, 2013, 12:50.
[3] Agrawal NK, Kant S. Targeting inflammation in diabetes: newer therapeutic options[J]. World J Diabetes, 2014, 5(5):697-710.
[4] Fang Q, Wang J, Wang L, et al. Attenuation of inflammatory response by a novel chalcone protects kidney and heart from hyperglycemia-induced injuries in type 1 diabetic mice[J]. Toxicol Appl Pharmacol, 2015, 288(2): 179-191.
[5] Luedde M, Lutz M, Carter N, et al. RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction[J]. Cardiovasc Res, 2014, 103(2):206-216.
[6] Liu ZY, Wu B, Guo YS, et al. Necrostatin-1 reduces intestinal inflammation and colitis-associated tumorigenesis in mice[J]. Am J Cancer Res, 2015, 5(10): 3174-3185.
[7] Eisen A, Fisman EZ, Rubenfire M, et al. Ischemic preconditioning: nearly two decades of research. A comprehensive review[J]. Atherosclerosis, 2004, 172(2):201-210.
[8] 孟繁學, 焦曉慧, 韓大英. ATP敏感鉀通道在心血管系統中的作用[J]. 中國病理生理雜志, 2000, 16(1): 94-96.
[9] Sierra A, Zhu Z, Sapay N, et al. Regulation of cardiac ATP-sensitive potassium channel surface expression by calcium/calmodulin-dependent protein kinase II[J]. J Biol Chem, 2013, 288(3):1568-1581.
[10]Zhao W, Zhang J, Lu Y, et al. The vasorelaxant effect of H2S as a novel endogenous gaseous KATPchannel opener[J]. EMBO J, 2001, 20(21): 6008-6016.
[11]Xu W, Wu W, Chen J,et al. Exogenous hydrogen sulfide protects H9c2 cardiac cells against high glucose-induced injury by inhibiting the activities of the p38 MAPK and ERK1/2 pathways[J]. Int J Mol Med,2013, 32(4):917-925.
[12]Xu W, Chen J, Lin J, et al. Exogenous H2S protects H9c2 cardiac cells against high glucose-induced injury and inflammation by inhibiting the activation of the NF-κB and IL-1β pathways[J]. Int J Mol Med, 2015, 35(1): 177-186.
[13]梁偉杰, 陳景福, 張穩柱, 等. ATP敏感性鉀通道在硫化氫抑制高糖引起的心肌細胞損傷中的作用[J]. 中國病理生理雜志, 2015, 31(5):785-790.
[14]梁偉杰, 何潔儀, 張穩柱, 等. 硫化氫通過抑制壞死性凋亡對抗高糖引起的H9c2心肌細胞損傷[J]. 中國病理生理雜志, 2016, 32(3):385-391.
[15]Huang CY, Kuo WT, Huang YC, et al. Resistance to hypoxia-induced necroptosis is conferred by glycolytic pyruvate scavenging of mitochondrial superoxide in colorectal cancer cells[J]. Cell Death Dis, 2013, 4:e622.
[16]Tay AS, Hu LF, Lu M, et al. Hydrogen sulfide protects neurons against hypoxic injury via stimulation of ATP-sensitive potassium channel/protein kinase C/extracellular signal-regulated kinase/heat shock protein 90 pathway[J]. Neuroscience, 2010, 167(2):277-286.
[17]Bian JS, Yong QC, Pan TT, et al. Role of hydrogen sulfide in the cardioprotection caused by ischemic preconditioning in the rat heart and cardiac myocytes[J]. J Pharmacol Exp Ther, 2006, 316(2):670-678.
(責任編輯: 林白霜, 羅 森)
Role of ATP-sensitive potassium channels in inhibitory effect of hydrogen sulfide on high glucose-induced inflammation mediated by necroptosis in H9c2 cardiac cells
LIANG Wei-jie1, 2, CHEN Mei-ji3, HE Jian-hua4, ZHANG Wen-zhu1, 2, CHENG Fei5, 6, LAN Jun5, 6, FENG Jian-qiang5, 6, HUANG Hui-min1, 2
(1DepartmentofCardiology,CentralHospitalofPanyuDistrict,2CardiovascularInstituteofPanyuDistrict,Guangzhou511400,China;3DepartmentofPediatrics,HuangpuDivision,TheFirstAffiliatedHospitalofSunYat-senUniversity,Guangzhou510700,China;4DepartmentofCardiology,ShiqiPeople’sHospitalofPanyuDistrict,Guangzhou511450,China;5DepartmentofCardiology,TheThirdPeople’sHospitalofDongguanCity,6CardiovascularInstituteofDongguanCity,Dongguan523326,China.E-mail: 9090430@qq.com)
AIM: To investigate the role of ATP-sensitive potassium (KATP) channels in the inhibitory effect of hydrogen sulfide (H2S) on high glucose(HG)-induced inflammation mediated by necroptosis in H9c2 cardiac cells.METHODS: The expression levels of receptor-interacting protein 3 (RIP3; an indicator of necroptosis) and cyclooxyge-nase-2 (COX-2) were determined by Western blot. The levels of interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) were detected by ELISA.RESULTS: After H9c2 cardiac cells were treated with 35 mmol/L glucose (HG) for 24 h, the expression of RIP3 was significantly increased. Pre-treatment of the cells with 100 μmol/L diazoxide (DZ; a KATPchannel opener) or 400 μmol/L NaHS (a donor of H2S) for 30 min considerably blocked the up-regulation of RIP3 induced by HG. Moreover, pre-treatment of the cells with 100 μmol/L 5-hydroxydecanoic acid (5-HD; a KATPchannel blocker) attenuated the inhibitory effect of NaHS on HG-induced up-regulation of RIP3. On the other hand, co-treatment of the cells with 100 μmol/L necrostatin-1 (a specific inhibitor of necroptosis) or pre-treatment of the cells with 100 μmol/L DZ or 400 μmol/L NaHS attenuated HG-induced inflammatory responses, evidenced by decreases in the expression of COX-2 and secretion levels of IL-1β and TNF-α. However, pre-treatment of the cells with 100 μmol/L 5-HD significantly attenuated the above anti-inflammatory effects of NaHS.CONCLUSION: KATPchannels play an important role in the inhibitory effect of H2S on HG-induced inflammation mediated by necroptosis in H9c2 cardiac cells.
ATP-sensitive potassium channel; Hydrogen sulfide; Necroptosis; High glucose; Cardiomyocyte; Inflammation
1000- 4718(2016)08- 1364- 06
2016- 04- 05
2016- 05- 06
廣東省自然科學基金資助項目(No. 2015A030313690);番禺區科技計劃項目(No. 2015-Z03-57)
R363
A
10.3969/j.issn.1000- 4718.2016.08.004
雜志網址: http://www.cjpp.net
△通訊作者 Tel: 020-34859342; E-mail: 9090430@qq.com
