膨脹單體在3D打印用光敏樹脂中的應用*
2016-06-05 07:48:03丁云雨林潤雄
化工科技 2016年2期
關鍵詞:質量
丁云雨,齊 迪,朱 軍,林潤雄
(青島科技大學 高性能聚合物研究院,山東 青島 266042)
3D打印是一種以數字文件為基礎,運用粉末狀金屬、塑料、液體光敏樹脂等可黏合材料,通過逐層打印的方式來構造物體的技術。3D打印技術廣泛應用于航空航天、醫療器械、產品設計等領域。按照所用材料的不同,3D打印技術可分為選擇性激光熔化成型技術(SLM)、選擇性激光燒結技術(SLS)、激光立體光刻技術(SLA)等[1],其中激光立體光刻(SLA)在3D打印技術中發展最早,最成熟,市場應用最廣泛。SLA所用材料為光敏樹脂,通過對光敏樹脂的逐層固化從而形成樣件[2]。在樹脂的固化過程中由于分子間鍵距發生變化,引起體積收縮。體積收縮輕則引發內應力加速試樣破壞,重則使樣件結構變形,翹曲,使成型失敗[3]。控制體積收縮率是制備3D打印用光敏樹脂的關鍵。
膨脹單體是一類在聚合反應過程中體積膨脹的物質[4]。自1972年Bailey發現膨脹單體以來,已有多種螺環原碳酸酯(SOC)、螺環原酸酯(SOE)、雙環原酸酯(BOE)被相繼合成出來[5]。膨脹單體因與環氧樹脂同為陽離子開環反應被廣泛應用于環氧樹脂的改性領域[6-7],然而將其運用到3D打印用光敏的文獻報道卻很少。
作者將一種合成的膨脹單體應用于3D打印用混雜型光敏樹脂中,研究了膨脹單體在混雜型光敏樹脂中的反應機理[9]以及對固化收縮率的影響,并測試了對光敏樹脂其它性能的影響,生成的膨脹單體在混雜型光敏樹脂中降低了其收縮率,取得了滿意的效果。
1 實驗部分
1.1 試劑與儀器
二正丁基氧化錫:工業級,南通艾德旺有限公司;三羥甲基丙烷:分析純,天津科密歐化學有限公司;甲苯、丙酮:分析純,煙臺三和化學試劑廠;正己烷:分析純,天津博迪化工股份有限公司;二硫化碳:分析純,成都市科龍化工有限公司;雙酚A型環氧樹脂621A-80、聚酯丙烯酸酯SF-319、脂肪族環氧樹脂UVR6105、三丙二醇二丙烯酸酯TPGDA:均為工業級,長興化工公司;引發劑184、引發劑UV6992:均為工業級,天津久日有限公司。
NEXUS 670型FT-IR紅外光譜儀:美國 Nicolet 公司;DRX500核磁共振儀:瑞士BRUKER公司;SPS-250固化儀:陜西恒通智能機器有限公司;GT-TCS-200型萬能拉力機:高鐵檢測儀器有限公司;數字式黏度計:上海尼潤智能科技有限公司。
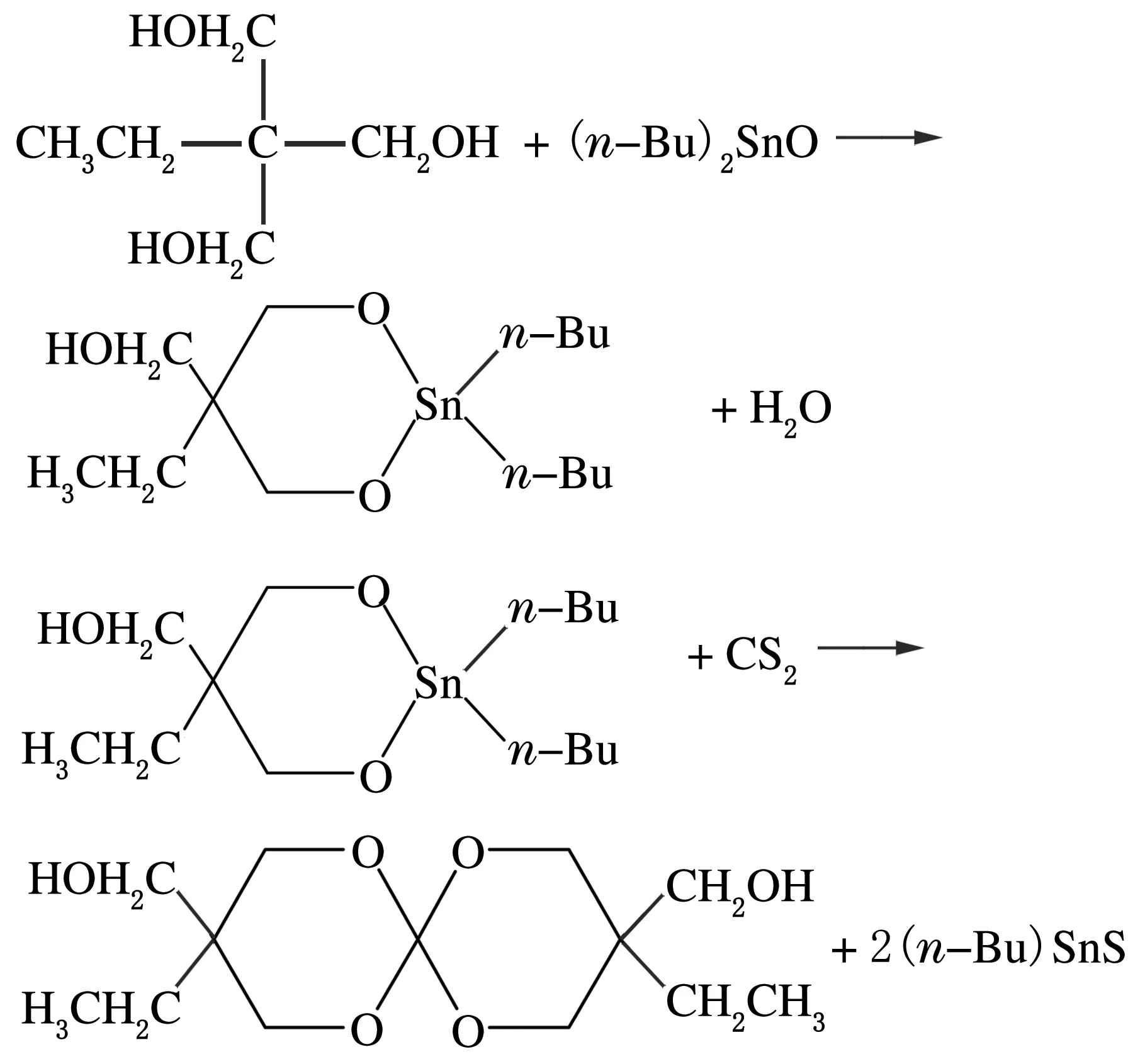
1.2 3,9二羥甲基-3′,9′二乙基-1,5,7,11-四氧螺雜[5,5]十一烷的合成
準確稱取24.9 g的二正丁基氧化錫加入到三口燒瓶中,加入150 mL的甲苯作為溶劑,再加入13.4 g 三羥甲基丙烷。燒瓶的一口裝上分水器,分水器上方加冷凝管,另外2個口分別裝溫度計和橡膠塞,磁子攪拌。升溫至120 ℃反應12 h或無水生成;冷卻至室溫,用恒壓漏斗滴加8 mL的CS2,滴加完畢后緩慢升溫至100 ℃攪拌回流反應12 h。減壓蒸餾出甲苯,所得黏稠液體用100 mL正己烷分5次洗滌,然后用35 mL甲苯加熱溶解,重結晶,真空干燥得到白色固體(產率70%)即為所得產物。單體的合成的反應式如下。
1.3 膨脹單體的均聚
將膨脹單體溶解到二氯乙烷中,m(膨脹單體)∶V(二氯乙烷)=1∶5 g/mL,再加入質量分數3%的三氟化硼乙胺作為催化劑,在30 ℃下攪拌反應24 h。反應結束后,將苯滴加到反應物中,使聚合物沉淀出來,產物經多次沉淀提純,干燥。
1.4 含膨脹單體光敏樹脂的制備
分別將質量分數為2.5%、5%、7.5%、10%、12.5%、15%、20%的膨脹單體加入到光敏樹脂的基本配方(見表1)中,用玻璃棒攪拌至均勻。在燒杯中加入磁子,溫度設定為35 ℃,控溫攪拌1 h,在35 ℃的真空烘箱中抽真空,放置于黑暗處靜置待用。

表1 混雜體系固化基本配方
1.5 表征與測試
1.5.1 紅外及核磁表征
將膨脹單體、膨脹單體均聚物以及含質量分數10%膨脹單體光敏樹脂固化前后物在FT-IR紅外光譜儀上采用全反射的方法進行譜圖測定,膨脹單體在DRX500核磁共振儀上采用氘代氯仿為溶劑進行譜圖測定。
1.5.2 光敏樹脂收縮率的測定
液體光敏樹脂的密度采用比重瓶法測定,固體密度參照國標GB 1463中的浮力法進行測量,光敏樹脂的總體積收縮率根據以下公式計算求得。
式中,V為光敏樹脂總體積收縮率;ρ0為固化前樹脂系統密度;ρc為固化后樹脂澆注體密度。
1.5.3 凝膠含量的測定
用SPS-250固化機對不同光敏樹脂進行單層固化,將固化好的薄片稱重,質量為m1,然后將薄片放入索氏提取器中用丙酮進行提取,10 h后或者其質量不變時取出薄片干燥,其質量為m2,w(凝膠)=m2/m1
1.5.4 力學性能的測定
用所配光敏樹脂為原料,利用SPS-250固化機制備拉伸試樣條,將拉伸試樣條在紫外燈下再固化后在萬能拉力機上進行拉伸測試。
2 結果與討論
2.1 膨脹單體結構討論
膨脹單體的紅外光譜圖和核磁共振氫譜圖見圖1、圖2。
由圖1可以看出在波數約1 220 cm-1時[10],產物有明顯的螺旋基團特征吸收峰,3 400 cm-1為寬的羥基吸收峰,2 850 cm-1為碳氫伸縮震動峰。由圖2可知,在0.82~0.86處為甲基質子峰(6H),1.33~1.37處為乙基上的亞甲基質子峰(4H),1.86處為羥基質子峰(2H),3.6~3.8處為與O相連的亞甲基質子峰。核磁峰積分面積之比與結構式基本相符,其它峰面積較小忽略不計。
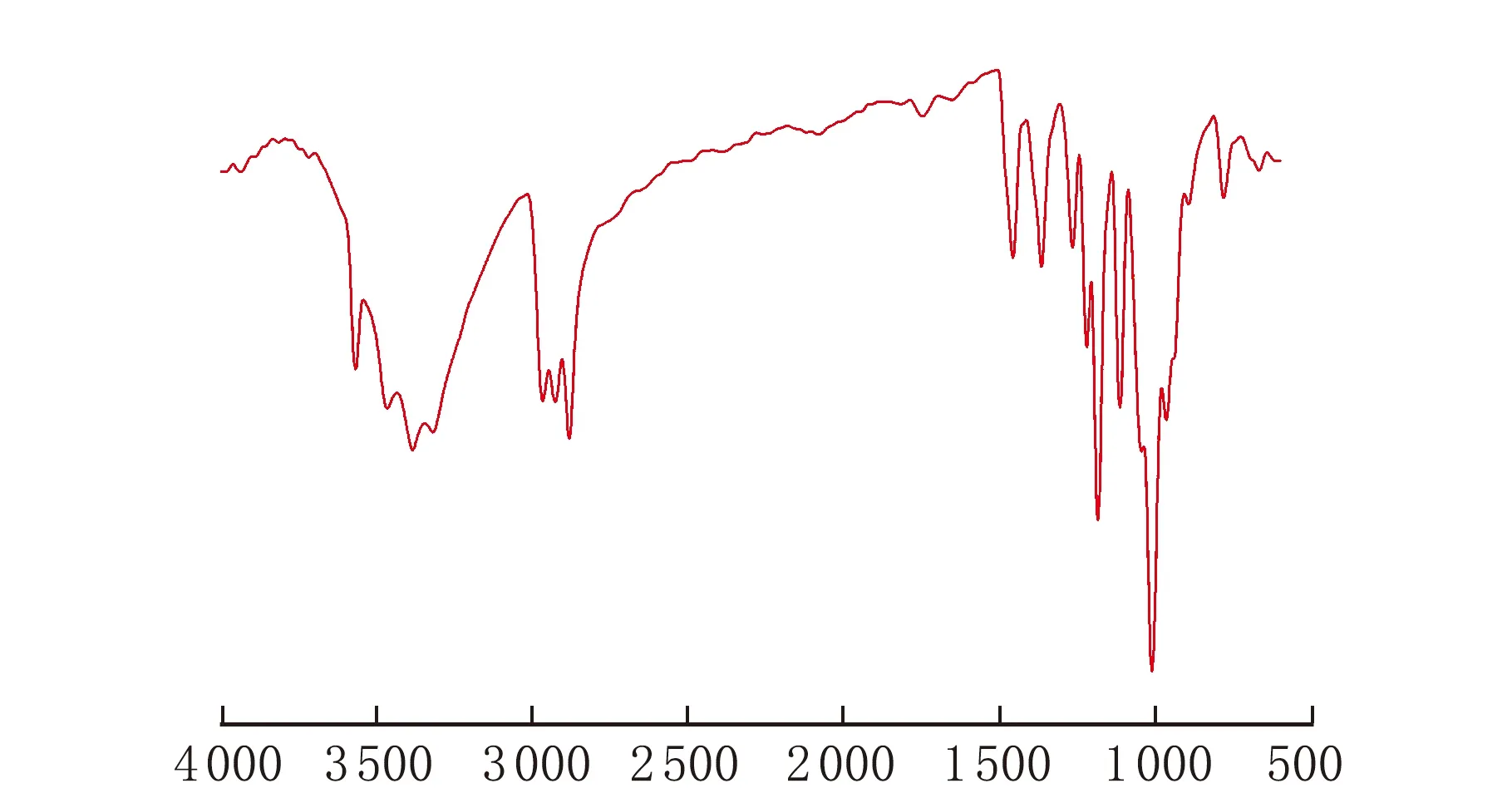
σ/cm-1圖1 膨脹單體的紅外光譜圖
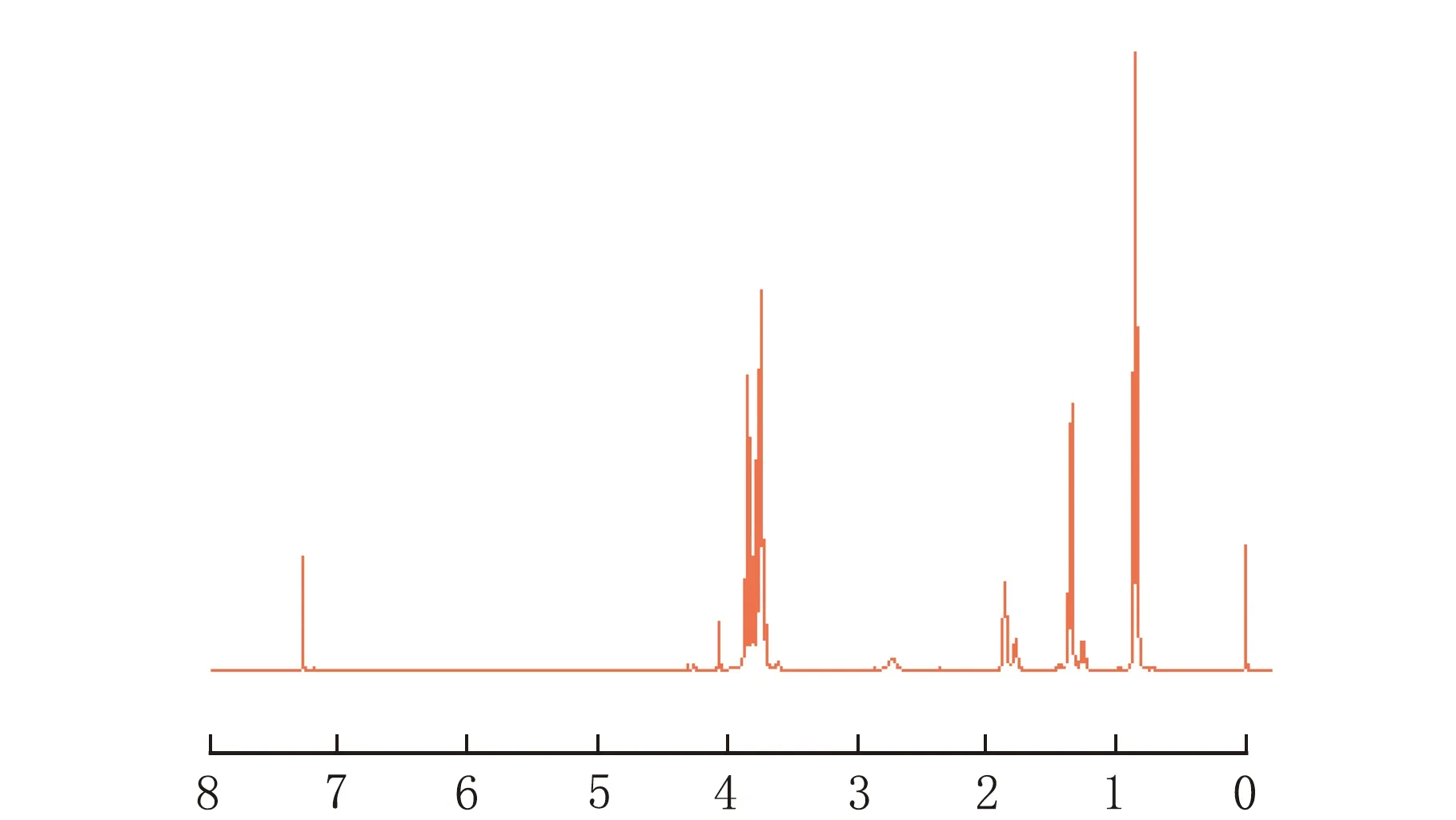
δ圖2 膨脹單體的核磁共振圖
由圖1和圖2證實了所合成的產物與1.2節中的反應式的最終產物結構相符,即產物為合成的膨脹單體。
2.2 膨脹單體的反應機理討論
膨脹單體均聚的紅外表征圖見圖3。從圖3中可以看出在1 220 cm-1的螺旋基團特征峰消失,同時在1 060 cm-1與1 250 cm-1處鏈狀醚鍵峰得到加強,在波數1 740 cm-1附近出現了碳酸酯羰基的強吸收峰。
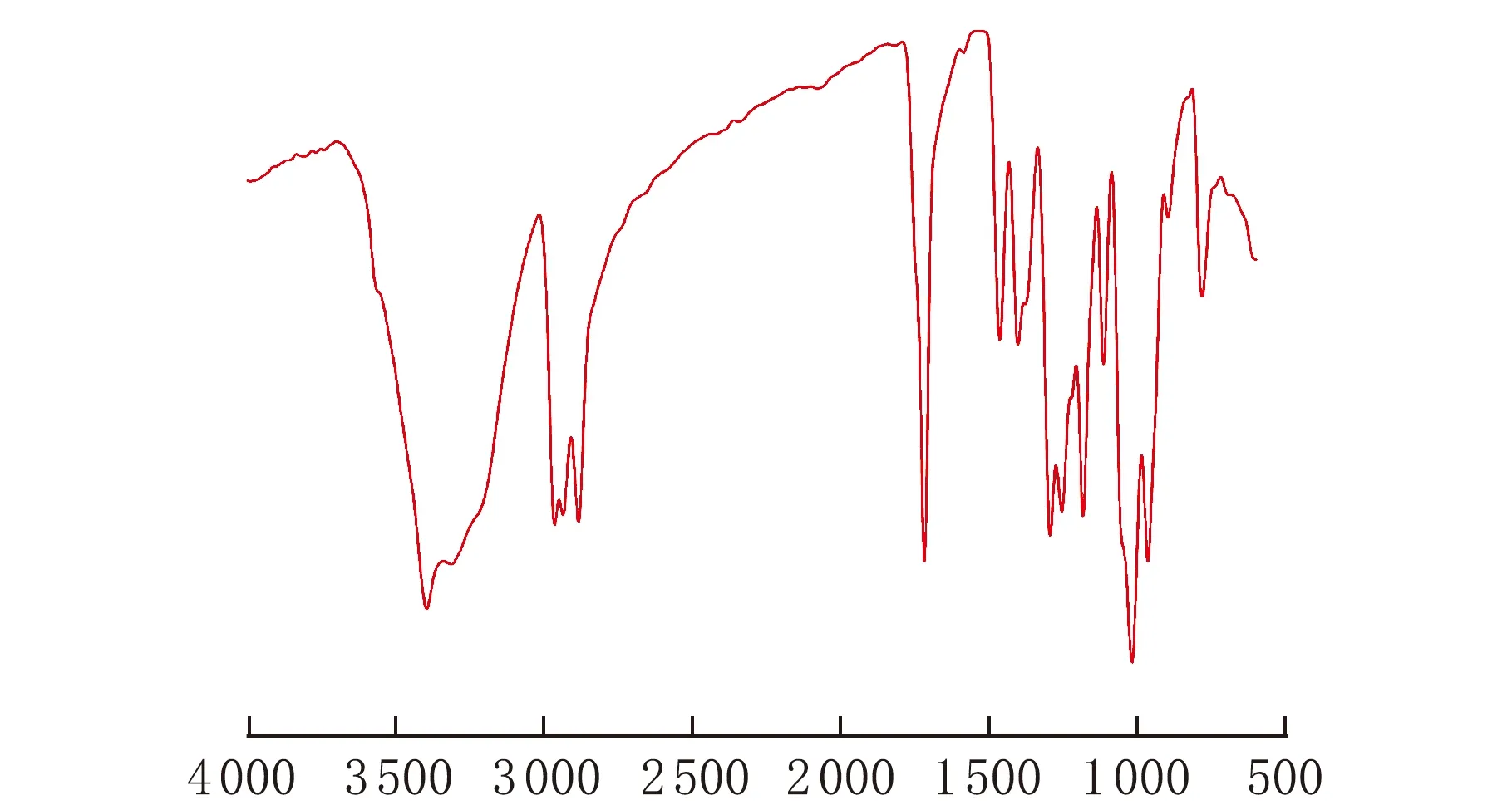
σ/cm-1圖3 均聚物的紅外表征
由圖3可知,膨脹單體在均聚過程中發生了雙開環反應,從側面也證明了2.1中所合成的產物為膨脹單體。雙開環反應歷程如下。
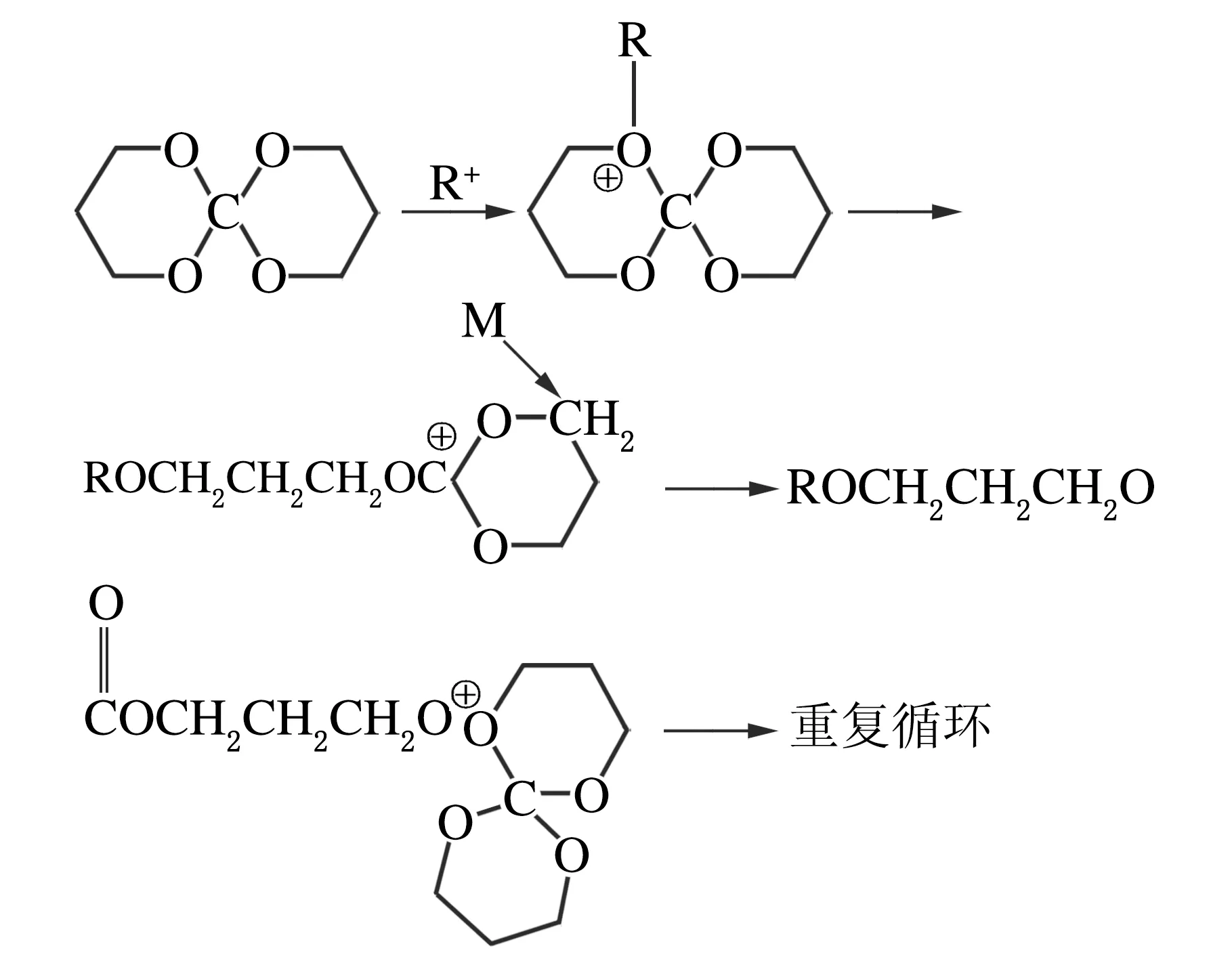
2.3 膨脹單體在光敏樹脂中固化的討論
對含質量分數10%膨脹單體光敏樹脂固化前后的紅外測試結果見圖4。
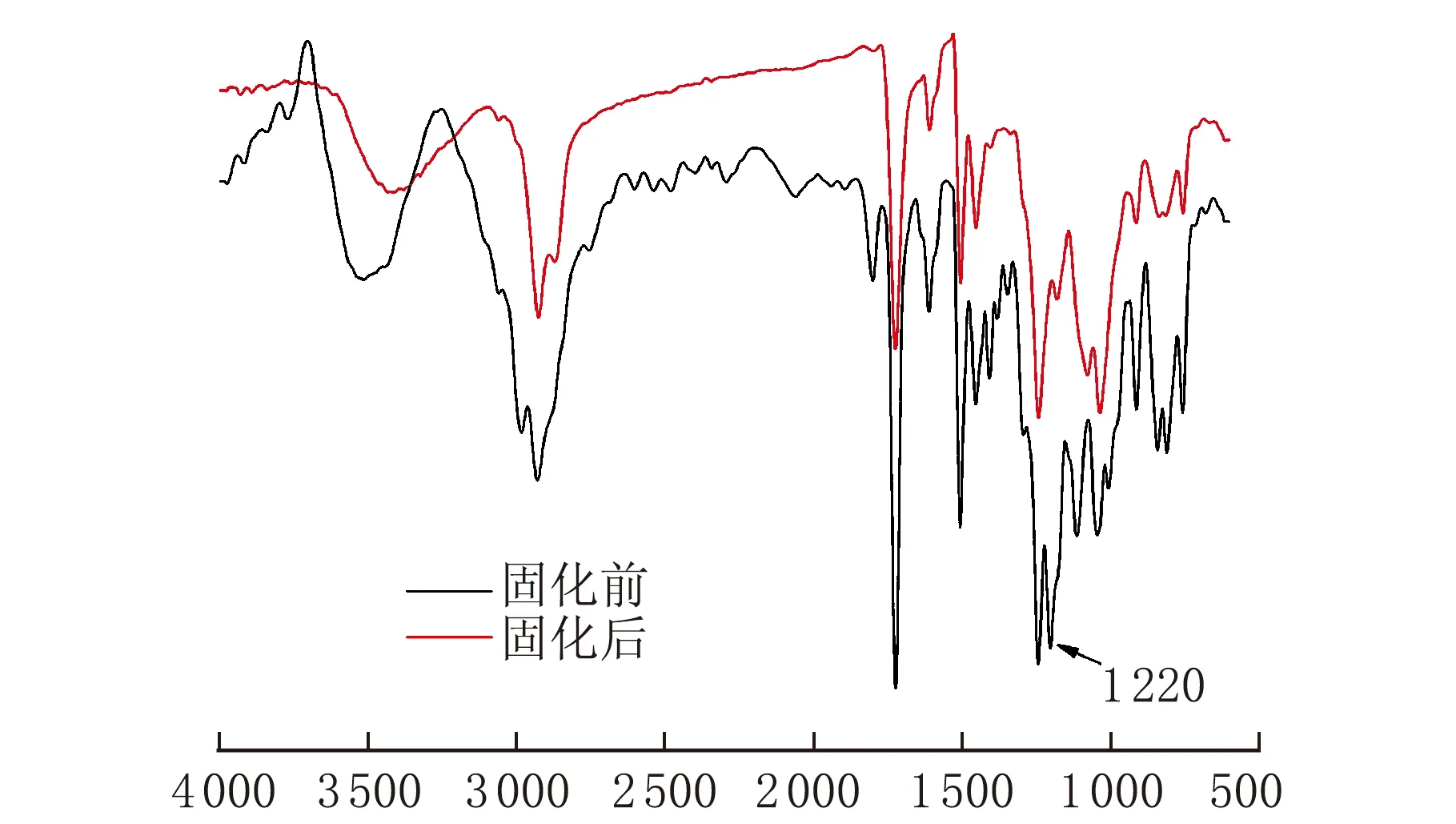
σ/cm-1圖4 光敏樹脂的紅外光譜圖
通過對比兩者紅外譜圖可以看出,固化后1 220 cm-1的螺旋基團的特征吸收峰消失,結合2.2中討論的膨脹單體均聚,可以得出在陽離子引發劑的作用下膨脹單體在光敏樹脂中發生了開環聚合反應。
2.4 膨脹單體對光敏樹脂性能影響的討論
2.4.1 膨脹單體對收縮率的影響
膨脹單體對收縮率的影響見圖5。
由圖5可以看出隨著膨脹單體含量的增加,樹脂的收縮率下降,但是在質量分數10%以后下降速率變慢。主要原因是在固化成型過程中,激光器的功率是一定的,而混雜型引發體系中既含有陽離子引發體系又含有自由基型引發體系,自由基的引發速度快,使固化過程中前期黏度急速上升,限制了膨脹單體的固化,其次相對于環氧樹脂的三元環來說,膨脹單體為雙六元環,鍵角大,環張力小,開環反應活性較小。因此當添加量超過質量分數10%以后,用于引發膨脹單體的能量減少。限制了膨脹單體的快速固化。基于上述原因在添加量超過質量分數10%后,一部分膨脹單體并沒有發生開環聚合,而是以單體的形式存在于固化后的樹脂中。
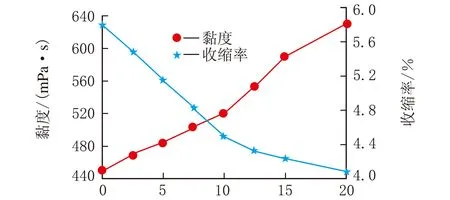
w(膨脹單體)/%圖5 膨脹單體與體積收縮率關系
樹脂的黏度隨著膨脹單體的加入緩慢上升,當含量超過質量分數10%以后黏度急劇升高而3D打印用光敏樹脂需要低黏度。綜合考慮在3D打印光敏樹脂中對膨脹單體的加入量應控制到質量分數10%以內。
2.4.2 膨脹單體對其它性能的影響
w(凝膠)可以反映樹脂由液態轉化為固態的轉化率。若膨脹單體參與反應則w(凝膠)會上升,反之則會下降。翹曲不僅與收縮率有關還與樹脂固化后的強度有關,高的力學強度可以大大降低發生翹曲的概率。實驗測定了含有膨脹單體光敏樹脂的w(凝膠)和拉伸強度,結果見圖6。
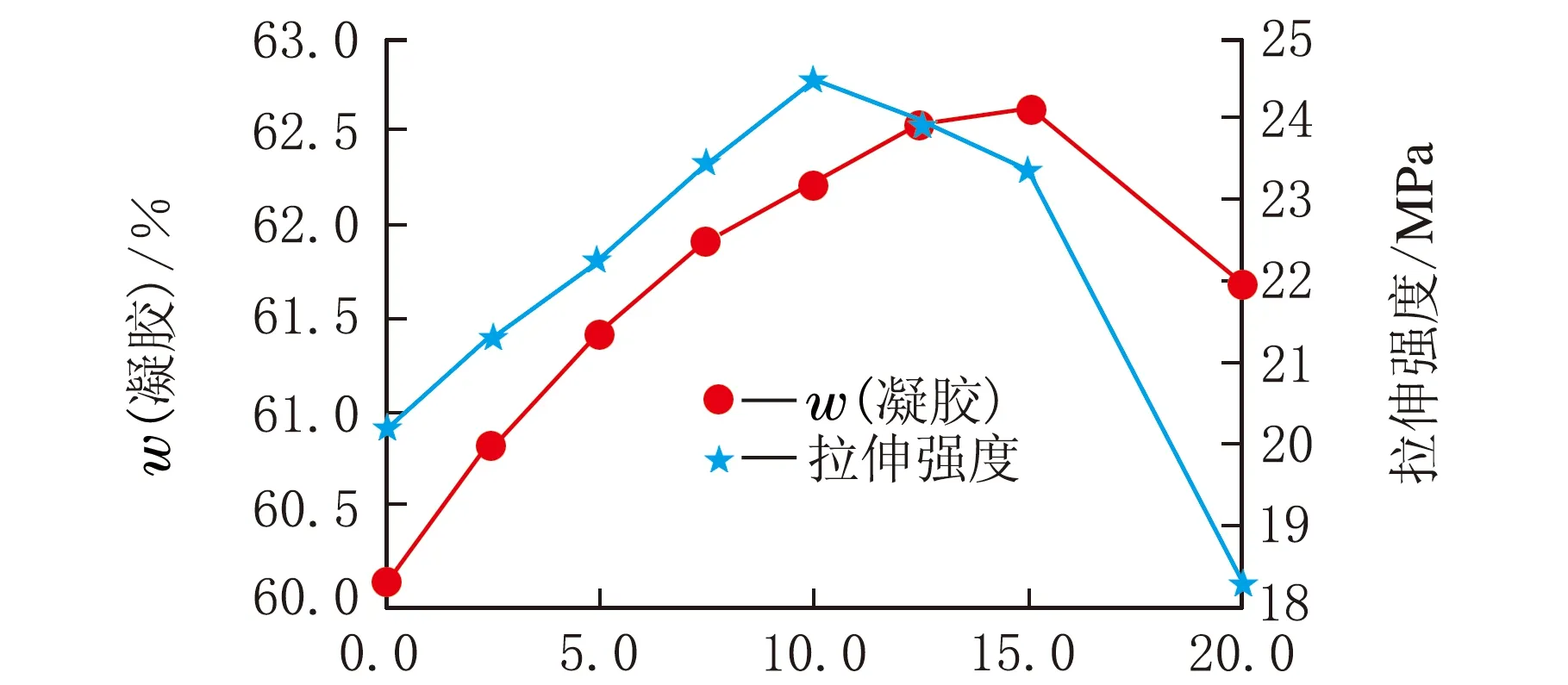
w(膨脹單體)/%圖6 w(膨脹單體)與w(凝膠)和拉伸強度關系
由圖6可以看出在w(膨脹單體)≤15%時,隨著膨脹單體的增加,w(凝膠)是一個增加的趨勢。但是變化率是逐漸變小的,這說明隨著膨脹單體含量的增加,只有部分單體參與了開環反應。當膨脹單體增加到質量分數20%時w(凝膠)變為下降趨勢,表明此時添加的膨脹單體已經不能固化。
從圖6中w(膨脹單體)與拉伸強度的關系曲線可以發現,w(膨脹單體)≤10%以內時,拉伸強度隨w(膨脹單體)的增加而增加。此時力學性能的增加主要是因為膨脹單體的加入降低了樹脂收縮時產生的內應力。當添加量在質量分數10%~15%時,拉伸性能呈現下降趨勢,主要原因是膨脹單體的加入降低了樹脂中剛性鏈的密度,使拉伸強度下降。當w(膨脹單體)=15%~20%時,拉伸性能大幅度下降,結合w(凝膠)與w(膨脹單體)的關系曲線圖可知此時下降的主要原因是過多膨脹單體加入使凝膠含量下降從而引起的力學性能下降。
3 結 論
(1) 利用兩步法合成一種螺旋原碳酸酯膨脹單體,通過表征確定了產物結構;
(2) 將膨脹單體應用到光敏樹脂中,通過對比樹脂固化前后的紅外譜圖及均聚物紅外譜圖確定了膨脹單體在光敏樹脂中可以發生開環反應;
(3) 通過測定樹脂的收縮率、w(凝膠)、拉伸性能,確定了w(膨脹單體)=10%時綜合性能最優;
(4) 通過添加膨脹單體,很好的改善了光敏樹脂的體積收縮,取得了滿意的效果。
參 考 文 獻:
[1] 王青崗,顏永年,郭戈,等.立體光刻(SL)工藝研究[J].新技術新工藝,2004,5:42-44.
[2] 王位,陸亞林,楊卓如.三維快速成型打印機成型材料[J].鑄造技術,2012,33(1):104-107.
[3] CANELLIDIS V,GIANNATSIS J,DEDOUSSIS V.Efficient parts nesting schemes for improving stereolithography utilization[J].Computer-Aided Design,2013,45(5):875-886.
[4] XU X Q,ZHOU L,LIANG B,et al.Synthesis of copolymers containing double spiro orthocarbonate and used as anti-shrinkage additives in epoxy resin composite[J].Polymer-Plastics Technology and Engineering,2014,53(8):753-759.
[5] HAYAL BULBUI SONMEZ,FRED WUDL.Synthesis of polymers based on spiro-orthocarbonates[J].Polymer Preprints,2003,44(2):803.
[6] SUN HWA YOO,CHANG KEUN KIM.Synthesis of a novel spiro orthocarbonate containing bisphenola unit and its application to the dental composites[J].Macromolecular Research,2010,18(10):1013-1020.
[7] 王長松,周本濂.螺環原碳酸酯的預聚物與環氧樹脂的共聚研究[J].高分子材料科學與工程,2000,16(1):28-31.
[8] 張露,邱婷,邢曉東.兩種膨脹單體的合成及其對牙科修復樹脂的改性[J].化工進展,2014,33(4):988-992.
[9] 周偉,方俊,等.一種新型膨脹單體預聚物的制備及其改性BDM/DABPA共混體的研究[J].玻璃鋼/復合材料,2013,4:14-17.
[10] 袁金穎,潘才元.膨脹單體對環氧樹脂的改性研究[J].功能高分子學報,1999,12(1):109-114.
猜你喜歡
中學生數理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(2018年6期)2018-04-22 03:16:54
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
