HPLC法測定勞拉西泮片有關物質的含量
2016-06-27 02:39:14孫愛華胡文輝
安徽醫藥 2016年5期
孫愛華,胡文輝
(1.黃岡市中心醫院藥劑科,湖北 黃岡 438000;2.鄂州市中心血站,湖北 鄂州 436000)
HPLC法測定勞拉西泮片有關物質的含量
孫愛華1,胡文輝2
(1.黃岡市中心醫院藥劑科,湖北 黃岡438000;2.鄂州市中心血站,湖北 鄂州436000)
摘要:目的建立勞拉西泮片的含量及有關物質的HPLC方法。方法以十八烷基硅烷鍵合硅膠作為填充劑的ZORBAX SB-C18柱(250 mm×4.6 mm,5 μm)為色譜柱,流動相:0.05 mol·L-1的磷酸二氫銨(含0.5%的三乙胺,用磷酸調pH至2.5)∶甲醇∶乙腈=35∶35∶30;流速:1.0 mL·min-1;檢測波長:235 nm;進樣量:10 μL;柱溫:30℃。結果勞拉西泮峰及各雜質峰均能良好分離。雜質A濃度與峰面積在1.5 μg·L-1~8.048 mg·L-1內線性關系良好。雜質B濃度與峰面積在15.0 μg·L-1~8.224 mg·L-1內線性關系良好。雜質C濃度與峰面積在1.7 μg·L-1~8.144 mg·L-1內線性關系良好;雜質D濃度與峰面積在2.5 μg·L-1~8.032 mg·L-1內線性關系良好;雜質E濃度與峰面積在3.0 μg·L-1~8.032 mg·L-1內線性關系良好。測定樣品含量平均值為97.9%,有關物質均符合要求。結論該方法快速、簡單,穩定,重現性好,可作為勞拉西泮片的含量及有關物質的檢測方法。
關鍵詞:勞拉西泮;色譜法,高壓液相
《美國藥典》35版[4]均收錄了勞拉西泮原料和其片劑、口服溶液劑及注射劑。兩版藥典中的勞拉西泮原料藥部分,列出了5個有關物質,有兩個工藝雜質,分別是雜質A和雜質B,其中雜質B即是工藝雜質又是降解產物,所以對雜質B進行了嚴格控制,對雜質A則未做具體要求。《歐洲藥典》[5]只收載了勞拉西泮原料藥,并用薄層色譜法對有關物質雜質A和雜質B進行測定,但未做具體要求。《中國藥典》2015版[6]均收載了勞拉西泮和勞拉西泮片,原料藥考察了雜質Ⅰ(即雜質B)和雜質Ⅱ(即雜質C),片劑只考察了雜質Ⅱ(即雜質C)。
為了提高勞拉西泮原料藥及制劑的質量,根據其生產工藝條件及降解特點,并結合強制破壞試驗的結果,最終確定了勞拉西泮原料藥及制劑的有關物質,包括雜質A、雜質B、雜質C、雜質D和雜質E(結構見圖1)。本文主要建立了一種可以同時測定勞拉西泮片劑和其有關物質檢測的HPLC方法,經驗證該方法操作簡便、靈敏和專屬性強。
1儀器與試藥
Agilent 1260型高效液相色譜儀,紫外檢測器(Agilent)及安捷倫色譜工作站,電子分析天平(AE230S 梅特勒-托利多上海有限公司),SB5200超聲儀(必能信超聲上海公司)。色譜純乙腈,色譜純甲醇,超純水,磷酸二氫銨(分析純,天津市科密歐化學試劑有限公司),三乙胺(分析純,國藥集團化學試劑有限公司),磷酸(分析純,國藥集團化學試劑有限公司)。勞拉西泮對照品(中國藥品生物制品檢定所,批號171253-200401),雜質A、B、C、D、E為自制。勞拉西泮片(泰國大西洋制藥有限公司,批號:140524),規格1.0 mg。
2方法和結果
2.1色譜條件色譜柱:ZORBAX SB-C18柱(250 mm×4.6 mm,5 μm);流動相:0.05 mol·L-1的磷酸二氫銨(含0.5%的三乙胺,用磷酸調pH至2.5)∶甲醇∶乙腈=35∶35∶30;流速:1.0 mL·min-1;檢測波長:235 nm;進樣量:10 μL;柱溫:30℃。
2.2溶液配制取勞拉西泮片對照品20片,研細,取細粉適量(約相當于勞拉西泮20 mg),置于100 mL容量瓶中,加入適量的流動相溶解,超聲10 min,放冷,補加流動相至刻度,搖勻,過濾,濾液作為供試品溶液。精密量取1.0 mL,置50 mL量瓶中,用流動相稀釋至刻度,搖勻,作為對照品溶液。
精密稱取勞拉西泮對照品5.06 mg、雜質A對照品5.03 mg、雜質B對照品5.14 mg、雜質C對照品5.09 mg、雜質D對照品4.97 mg、雜質E對照品5.02 mg,分別置于25 mL容量瓶中,加入乙腈溶解并稀釋至刻度,搖勻,作為勞拉西泮和各雜質對照品的貯備溶液。
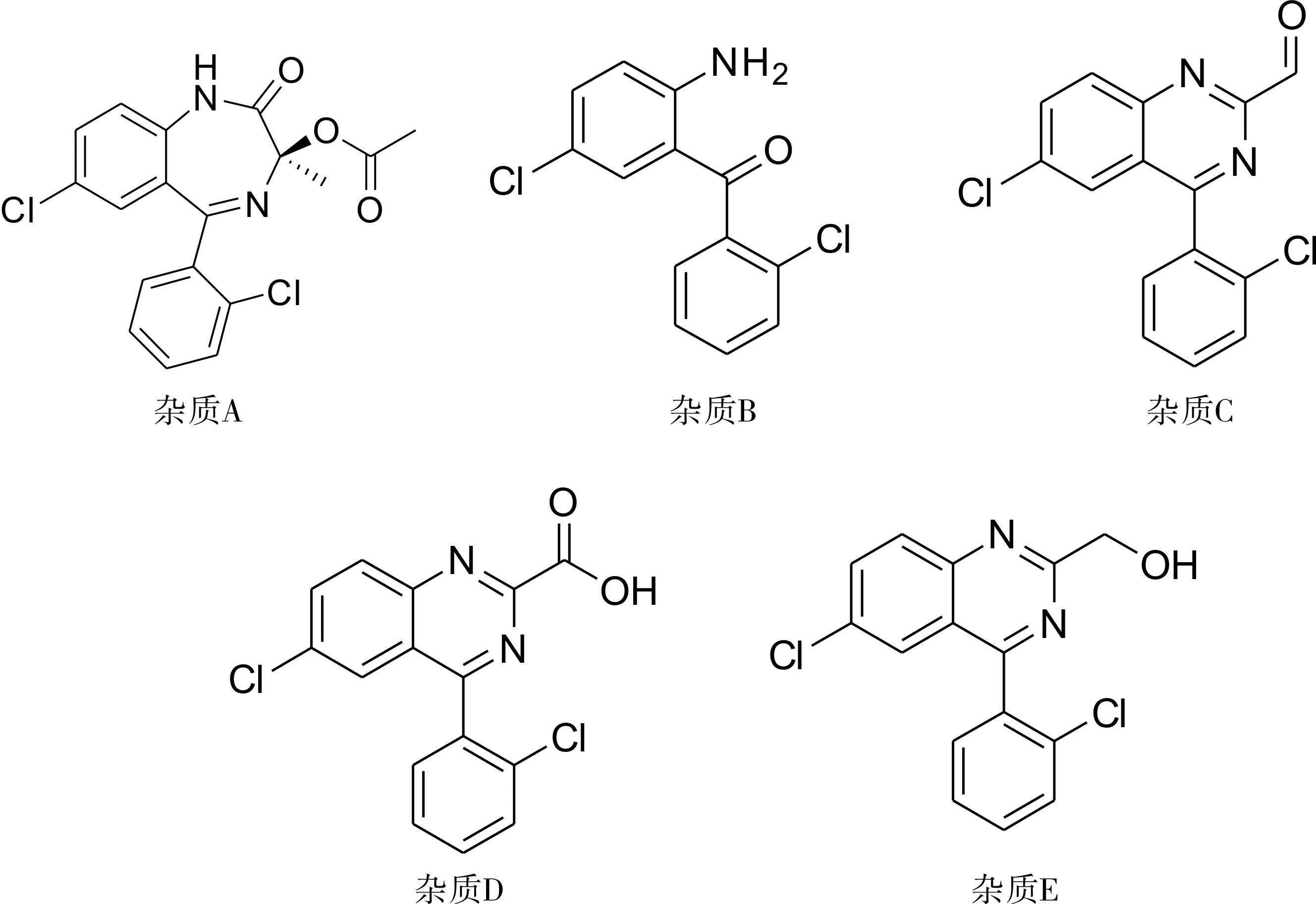
注:雜質A.3(RS)-7-氯-5-(2-氯苯基)-1,3-二氫-乙酰氧基-2H-苯二氮-2-酮;雜質B.(2-氨基-5-氯苯基)(2-氯苯基)甲酮;雜質C.6-氯-4-(2-氯苯基)-喹唑啉-2-醛;雜質D.6-氯-4-(2-氯苯基)-喹唑啉甲酸;雜質E.6-氯-4-(2-氯苯基)-喹唑啉甲醇。
圖1勞拉西泮原料藥及制劑的有關物質結構圖
2.3方法學考察
2.3.1系統適用性試驗分別精密量取上述勞拉西泮及各雜質對照品的貯備溶液1.0 mL,置于10 mL的容量瓶中,加入流動相溶解并稀釋至刻度,作為系統適用性溶液。精密量取10 μL注入高效液相色譜儀,記錄色譜圖,各峰理論板數要求不低于3 000,分離度不低于1.5,如圖2所示。

注:1.勞拉西泮;2.雜質A;3.雜質D;4.雜質E;5.雜質C;6.雜質B。
圖2系統適應性試驗圖譜
2.3.2檢測限及定量限測定取配制的各雜質對照品溶液1.0 mL,分別置于100 mL容量瓶中,加入流動相稀釋至刻度,搖勻,作為檢測限定量限的試驗溶液, 按照上述色譜條件進樣,進樣10 μL,根據信噪比法進行試驗,信噪比為3計算檢測限,信噪比為10計算定量限,結果見表1。
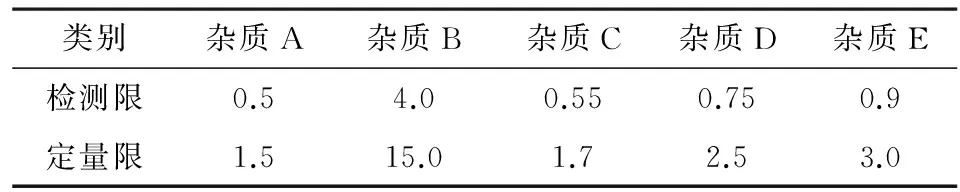
表1 各雜質檢測限及定量限/ng
2.3.3線性試驗精密稱量各雜質對照品的貯備溶液0.5、1.0、1.5、2.0 mL,分別于50 mL容量瓶中,用流動相稀釋至刻度,搖勻,作為各雜質對照品的定量溶液,分別進樣10 μL,記錄色譜圖。以峰面積為縱坐標(A),以濃度(mg·L-1)為橫坐標(C),得各雜質的回歸方程及線性范圍,結果見表2。
2.3.4精密度試驗精密量取系統適用性溶液10 μL,重復進樣6次,分別計算各峰面積的相對標準偏差。勞拉西泮和雜質A、B、C、D、E的峰面積相對標準偏差分別為0.27%、0.39%、1.04%、1.54%、0.86%、1.02%(n=6)。
2.3.5溶液穩定性試驗分別精密量取系統適用性溶液10 μL,于0、2、4、8、12 h進行測定,勞拉西泮及雜質A、B、C、D、E峰面積的相對標準偏差分別為0.92%、0.83%、1.04%、0.78%、1.16%、1.28%,說明樣品及各個雜質在12 h內穩定。
2.3.6回收率試驗精密稱取勞拉西泮對照品10.03、10.11、10.05 mg,分別置50 mL量瓶中,精密量取各雜質對照品貯備液2.5、5.0、7.5 mL,置上述量瓶中,加入流動相稀釋至刻度,搖勻,分別作為加樣回收率的低、中、高濃度。根據各雜質峰面積計算其回收率,結果見表3。
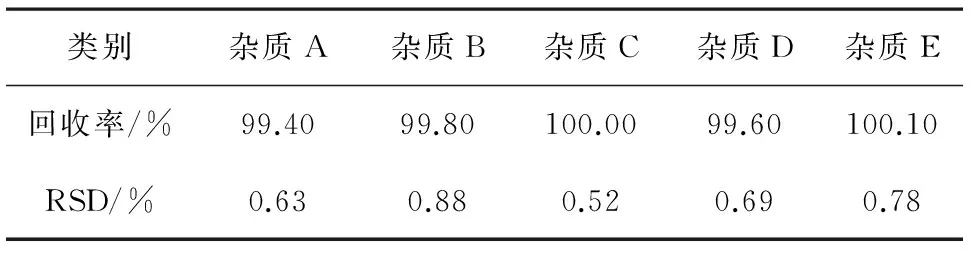
表3 各雜質回收率及RSD結果匯總
2.3.7專屬性試驗精密稱定勞拉西泮對照品100 mg于50 mL的容量瓶中,加入適量甲醇振搖溶解,用甲醇稀釋至刻度。量取5.0 mL對照品溶液于25 mL的容量瓶中,加入0.1 mol·L-1鹽酸溶液5.0 mL搖勻,甲醇稀釋至刻度,定容,置于100℃水浴鍋中4 h,放冷備用。量取5.0 mL對照品溶液,加入0.1 mol·L-1氫氧化鈉搖勻,甲醇稀釋至刻度,定容,置于100℃水浴鍋中4 h,放冷備用。量取5.0 mL對照品溶液,加入30%雙氧水溶液5.0 mL,搖勻,室溫放置24 h。另外分別量取5.0 mL的溶液2份,置于25 mL的容量瓶中,加入甲醇稀釋至刻度。一個置于100℃水浴鍋中加熱4 h,放冷。另外一個置于強光(5 000 Lx)下照射12 h后,放冷。分別精密量取上述各破壞項中的溶液1.0 mL,加入流動相稀釋至10.0 mL,按照上述色譜條件取10 μL進樣測定,記錄色譜圖,見圖3。
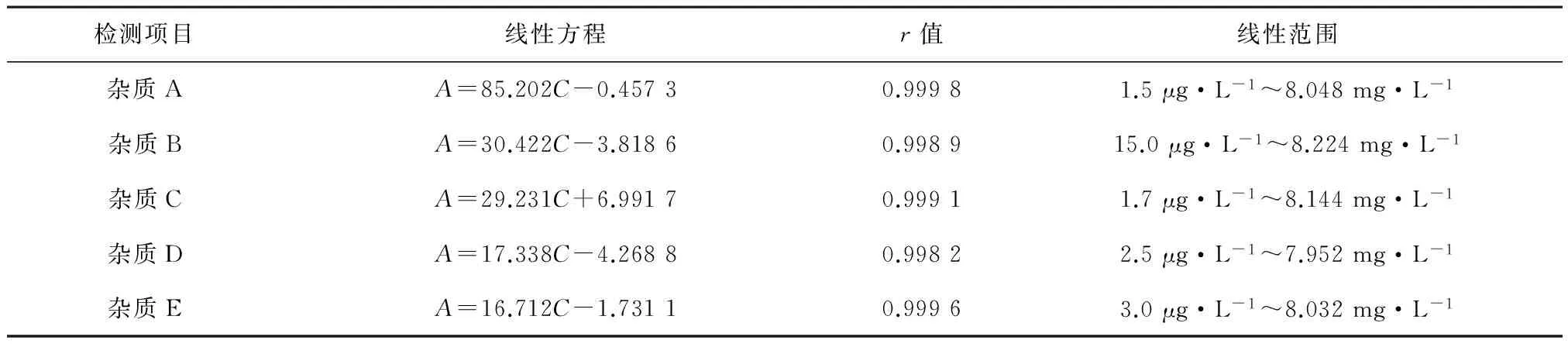
表2 各雜質線性方程及線性范圍

圖3 專屬性試驗圖譜
通過圖譜可以看出,勞拉西泮在堿性條件及光照條件下較為穩定,未見明顯雜質出現。在酸性條件下破壞出雜質B,峰面積按照面積歸一化計算約為17.6%。在氧化破壞中產生雜質C、雜質D及雜質E,按照面積歸一化法計算峰面積分別為2.5%、3.1%、2.7%。高溫破壞雜質主要為雜質C,峰面積按照面積歸一法計算為5.1%。
2.4樣品含量測定取上市品勞拉西泮片,研細,精密稱量粉末適量(相當于勞拉西泮10 mg),置于50 mL容量瓶中,加入甲醇適量,超聲溶解10 min,補加流動相稀釋至刻度,搖勻,過濾,濾液作為供試品備用。按照上述色譜條件,精密量取10 μL進色譜儀,記錄圖譜。已知雜質按照雜質對照計算,未知雜質按照0.5%勞拉西泮的自身對照法來計算樣品含量及有關物質含量。檢查結果表明樣品中雜質B和雜質E均未檢出,其余有關物質和含量測定結果見表4。
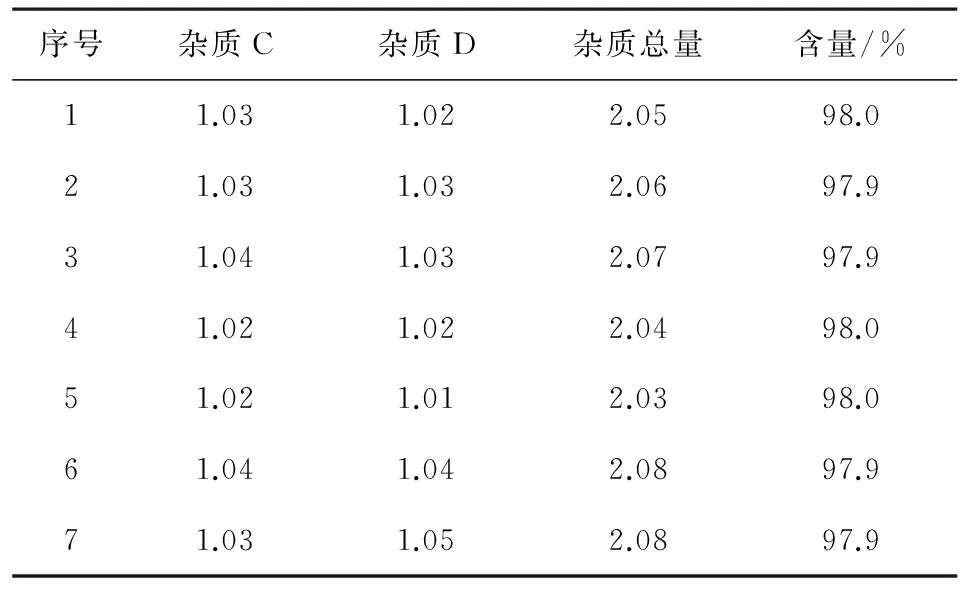
表4 勞拉西泮片有關物質及含量測定結果
3討論
勞拉西泮在230 nm處有最大吸收,綜合降解產物及各有關物質的性質,選擇235 nm為有關物質及含量的檢測波長,此波長可以兼顧其他各有關物質的檢測。試驗過程中分別試驗了甲醇和磷酸緩沖鹽、乙腈和磷酸緩沖鹽等不同流動相,發現單一有機溶劑和磷酸緩沖鹽的組合不能將勞拉西泮和各個有關物質有效分類。將甲醇和乙腈同等比例再與磷酸緩沖鹽混合可以達到有效分離各個有關物質及勞拉西泮。由于雜質D為一極性較大的羧酸類化合物,保留時間受pH的影響較大。故試驗中采用了磷酸鹽緩沖液系統,且用磷酸調節pH為2.5。
參考文獻
[1]孫定人,張石革,梁之江.國家臨床新藥集[M].北京:中國醫藥科技出版社,2001:19.
[2]郜娜,賈琳靜,謝敏,等.反相高效液相色譜法測定人血漿中勞拉西泮的濃度[J].藥物分析雜志,2005,25(4):423-425.
[3]程東升,陳蕾,南楠.勞拉西泮原料及其片劑中有關物質的檢查[J].藥物分析雜志,2006,26(12):1800-1803.
[4]The United States Pharmacopeial Convention US.Pharmacopeia (35)/ National Formulary(30)[S],2011:3720.
[5]European Directorate for the Quality Control of Medicines.European Pharmacopoeia 8.0[S],2013:1931.
[6]國家藥典委員會.中國藥典(二部)[S].北京:中國醫藥科技出版社,2015:452.
HPLC determination of the related substances in iorazepam tablets
SUN Ai-hua1,HU Wen-hui2
(1.PharmaceuticalDepartment,HuanggangCentralHospital,Huanggang,Hubei438000,China;2.EzhouCentralBloodStation,Hubei436000,China)
Abstract:ObjectiveTo determine the related substances and content in iorazepam tablets by HPLC.MethodsThe chromatographic separation was performed on ZORBAX SB-C18column(4.6 mm×250 mm,5 μm)that was made by alkyl-bonded silica gels.The mobile phase was 0.05 mol·L-1ammonium dihydrogen orthophosphate (including 0.5% trimethylamine and pH adjusted to 2.5 with phosphoric acid)∶methanol∶acetonitrile=35∶35∶30.The flow rate was 1.0 mL·min-1,the detection wavelength was 235 nm,the sample size was 10 μL,and the column temperature was set at 30℃.ResultsThe resolution between iorazepam and the other peaks met the requirements.The linearity of related substance A concentration and peak area was 1.5 μg·L-1~8.048 mg·L-1.The linearity of related substance B concentration and peak area was 15.0 μg·L-1~8.224 mg·L-1.The linearity of related substance C concentration and peak area was 1.7 μg·L-1~8.144 mg·L-1.The linearity of related substance D concentration and peak area was 2.5 μg·L-1~7.952 mg·L-1.The linearity of related substance E concentration and peak area was 3.0 μg·L-1~8.032 mg·L-1.Determination of the sample average content was 97.9%,and the related substance met the requirements.ConclusionsThe method was quick,simple,stable,and well repeatable,which could be used to determine the content of iorazepam tablets and its related substances.
Key words:iorazepam;Chromatography,High Pressure Liquid
通信作者:胡文輝,男,副主任技師,研究方向:臨床檢驗與實驗診斷,E-mail:2531134237@qq.com
doi:10.3969/j.issn.1009-6469.2016.05.014
(收稿日期:2015-10-15,修回日期:2016-02-23)
