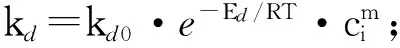長鏈烷烴脫氫制單烯烴動力學模型Ⅱ.工業脫氫反應器模擬
2016-07-05 07:35:51江洪波王雅純周立群
石油學報(石油加工) 2016年2期
江洪波, 王雅純, 張 晴, 周立群, 王 玉, 曹 晶
(1. 華東理工大學 石油加工研究所, 上海200237; 2. 中國石化 金陵石化有限責任公司烷基苯廠, 江蘇 南京210046)
?
長鏈烷烴脫氫制單烯烴動力學模型Ⅱ.工業脫氫反應器模擬
江洪波1, 王雅純1, 張晴1, 周立群2, 王玉2, 曹晶2
(1. 華東理工大學 石油加工研究所, 上海200237; 2. 中國石化 金陵石化有限責任公司烷基苯廠, 江蘇 南京210046)
摘要:以高純度的長鏈烷烴為原料,采用固定床工業脫氫反應器進行反應制取直鏈單烯烴,其中脫氫催化劑在使用過程中會緩慢失活。采用一維擬均相絕熱徑向反應器模型模擬工業脫氫反應器,結合長鏈烷烴的反應動力學模型,推導出物料衡算式以及熱量衡算式,模擬估算工業脫氫反應,還定量預測和分析了反應器操作條件對反應的影響,確定了工業反應器催化劑失活動力學方程。結果表明,模型擬合效果良好,催化劑的活性隨使用時間增長而下降,并成線性關系。該模型可用于催化劑活性變化的定量預測。
關鍵詞:長鏈烷烴; 脫氫; 徑向反應器; 失活; 模擬
烴的脫氫在有機合成中是重要的過程之一,其中長鏈烷烴脫氫制備直鏈單烯烴并合成直鏈烷基苯是合成洗滌劑和表面活性劑的重要原料[1]。目前,世界上89%的直鏈烷基苯都是由直鏈烷烴脫氫并與苯合成制得[2]。工業上,直鏈烷烴脫氫反應是在氫氣的存在下,在軸向或徑向活塞流絕熱反應器中進行,在0.2MPa、465~495℃的反應條件下,以鋁或沸石為載體的鉑催化劑作為催化劑,脫氫生成直鏈單烯烴。脫氫反應過程伴隨著異構化、裂化、二聚化等副反應,且產生的二烯烴和芳烴會降低催化劑活性,縮短循環使用次數,而催化劑活性、選擇性和穩定性都可影響直鏈烷基苯的生產成本和質量[3-4]。
近年來,長鏈烷烴脫氫研究不僅僅局限于催化劑的改善和開發,計算機輔助對于工業反應的操作優化的重要性日益顯現[5]。Alexis等[2]根據質量守恒和能量守恒定律,推導出滴流床反應器中C12~C14在PtCu催化劑催化下的脫氫反應模型,并通過實際操作工況檢驗了模型的適用性;Frantsina等[6]推導出工業上C9~C14直鏈烷烴在Pt脫氫催化劑催化下的脫氫反應模型,考察了原料組分的變化對于目標產物的影響,具有一定的適用性。國內對于直鏈烷烴脫氫的研究主要是關于催化劑性能的改進,對于脫氫反應動力學和工業反應器的模擬研究較少。
筆者采用一維擬均相絕熱反應器模型模擬工業脫氫反應器,結合已開發的長鏈烷烴的脫氫反應動力學模型[7],推導出物料衡算式和熱量衡算式,采用Runge-Kutta法求解方程組,確定床層中濃度分布和溫度分布,以及催化劑活性隨時間變化的規律,并與工業反應器結果比較,確定了模型的適用性。
1長鏈烷烴催化脫氫制取烷基苯工藝流程
從分子篩脫蠟裝置來的新鮮輕蠟與從烷基化裝置出來的循環烷烴混合后一起進入物料平衡罐。從平衡罐出來的混合物料與循環氫氣以大約1∶5的摩爾比進入換熱器,經加熱爐加熱到指定溫度后進入徑向固定床脫氫反應器,在脫氫催化劑作用和臨氫狀態下進行脫氫反應。脫氫催化劑在使用過程中會緩慢失活,當催化劑活性下降到一定程度時置換或再生催化劑,因而需要兩臺脫氫反應器切換操作,一開一備。從脫氫反應器出來的脫氫物料經分離罐將氫氣和烷、烯烴分離,分離后的烷、烯烴進入雙烯選擇加氫反應器進行選擇性加氫,然后進入烷基化裝置進行烷基化反應,未反應的烷烴作循環烷烴使用。
針對以長鏈烷烴為原料脫氫制取烷基苯工藝流程中脫氫部分工業反應器進行模擬優化。
2長鏈烷烴脫氫徑向反應器的數學模型
2.1長鏈烷烴脫氫反應動力學
長鏈烷烴催化脫氫原料是多種不同碳數直鏈烷烴的混合物。不同碳數直鏈烷烴的脫氫反應速率有所不同,整個脫氫反應網絡涉及到單烯烴、二烯烴、三烯烴、芳烴、裂化產物、焦炭等多種反應中間產物和最終產物,反應網絡比較復雜。工業數據分析表明,在長鏈烷烴脫氫體系中存在直鏈烷烴、直鏈單烯烴、非正構單烯烴、雙烯烴、裂解產物、芳烴和氫氣,具體的反應網絡如圖1所示,由此可以得到脫氫反應的表觀集總動力學模型[7]。但經過對比工業數據和實驗室反應結果發現,工業上脫氫反應所生成的二烯烴及芳烴的質量分數大于實驗室的結果,因此在對工業反應器進行模擬計算之前,對轉化生成二烯烴及芳烴的相關反應速率常數進行了適當修正。其中當溫度為488℃時,k2=10.671670L/(g·h),k10=4.505942L/(g·h),k11=4.446996L/(g·h),k12=8.519501L/(g·h),反應活化能保持不變。
2.2長鏈烷烴脫氫反應器模型
在本研究中,假定催化劑床層內流體為理想平推流,采用一維擬均相徑向反應器模型對工業脫氫反應器進行模擬優化。根據質量守恒和能量守恒定律,推導出長鏈烷烴脫氫體系的物料衡算式(1)~(7)和熱量衡算式(8)。
(1)
(2)
(3)
(4)
(5)
(6)
k6CmsCH2-k7CnsCH2-k-3CnsCH2+4k9Cps+3k10Cms+3k11Cns+k12Cns-k-12CdsCH2)·a
(7)
k-3CnsCH2ΔH-3+k4CnsΔH4-k5CpsCH2ΔH5-k6CmsCH2ΔH6-k7CnsCH2ΔH7+2k8CdsΔH8+4k9CpsΔH9+
3k10CmsΔH10+3k11CnsΔH11+k12CnsΔH12-k-12CdsCH2ΔH-12)·a/Cp
(8)
式(1)~(8)中,G為原料體積流率,m3/h;r為催化劑床層半徑,m;h為催化劑床層高度,m;Cps、Cms、Cns、Cds、C 方程式(1)~(8)構成了C9~C14直鏈烷烴脫氫徑向反應器的一維擬均相反應器模型。采用Runge-Kutta法求解出產物組成和物料溫度沿催化劑床層的徑向分布。 3模擬結果與分析 3.1模型計算結果和實驗結果的對比 在反應器進口溫度470~480℃、絕對壓力0.22~0.24MPa、氫/烴摩爾比5.7~6.9、液時空速23~26h-1的條件下,考察了長鏈烷烴C9~C14在金陵石化NDC-10脫氫催化劑催化下的脫氫反應,反應器進口混合物料組成列于表1。在催化劑使用的1個周期內,參照上述脫氫反應器模型,模擬計算反應器的出口物料組成、出口溫度和催化劑活性,結果示于 圖2。分析對比結果,所有物質的實際值與計算值的平均相對偏差為4.17%,其中正構烷烴的平均相對偏差為0.27%,單烯烴的平均相對偏差為0.14%。總的平均相對偏差的主要誤差來源于二烯烴、芳烴等副產物,它們的絕對誤差不大,但由于這些副產物產量較小,因而較小的絕對誤差也可能導致較大的相對偏差。考慮到工業實測數據本身存在一定誤差,可以接受所得實際值與計算值的平均相對偏差水平,反應器模型的計算結果可靠。 以催化劑使用周期為1個活性計算周期,考察在1個使用周期內活性隨時間的變化,結果示于圖3。分離失活動力學研究方法適用于因結焦或燒結而引起的失活過程,可用于直鏈烷烴脫氫催化劑的失活情況。根據分離失活動力學研究方法的假定[8],反應動力學不隨催化劑失活而變化,催化劑活性隨操作過程的變化可與反應動力學分開作獨立研究,脫氫催化劑的失活方程式如式(9)所示。 (9) 脫氫反應動力學為基于工業脫氫催化劑(相同顆粒大小)并在工業操作條件下建立的表觀反應動力學,無需考慮內、外擴散的影響,可直接用于工業反應器的模擬計算[7]。在催化劑的1個使用周期內,反應器操作條件變化范圍較小,反應原料進料組成基本不變,因而在考察周期內活性變化時,可以忽略其他因素對于催化劑活性的影響。因而脫氫催化劑的失活動力學方程式可表示為式(10)。 (10) 通過擬合曲線,確定催化劑的活性在整個脫氫工藝過程中與使用時間成線性關系,且滿足式(11): a=0.83151-9.30135×10-5t (11) 式(11)中t為催化劑使用時間,h。與實驗室所得催化劑失活規律略有不同,推測其原因主要有兩個:(1)為了避免催化劑活性下降過快而影響產物的產率,工業反應器采用了高于實驗室動力學實驗的反應壓力,并充入少量水蒸氣來減緩催化劑失活,抑制了整個操作周期內催化劑的失活速率;(2)工業反應器的入口溫度隨著操作時間延長逐淅升高,使得相同活性的催化劑失活速率有所加快。兩者共同作用導致催化劑的活性與操作時間成線性關系,從圖3 可以看出,催化劑活性隨使用時間的延長而逐漸降低,且降低的速度基本一致。 3.2反應條件對直鏈烷烴脫氫反應影響的模擬結果 3.2.1進口溫度的影響 在絕熱脫氫反應器絕對壓力0.24MPa、氫/烴摩爾比6.0、液時空速26h-1的條件下,改變反應器的進口溫度,假定催化劑處于同一活性水平(a=0.71),模擬計算結果示于圖4。從圖4可以看出,長鏈烷烴脫氫轉化率隨進口溫度的升高而升高,目標產物單烯烴的選擇性隨進口溫度的升高而降低。對可逆吸熱脫氫反應來說,反應溫度的升高有利于脫氫平衡,且加快脫氫主反應速率,但活化能比脫氫反應大的裂解等副反應的反應速率加快更甚,結果是反應的轉化率增加了,但單烯烴選擇性卻下降了。 3.2.2壓力的影響 在絕熱脫氫反應器進口溫度為480℃、氫/烴摩爾比6.0、液時空速26h-1的條件下,改變絕對壓力,假定催化劑處于同一活性水平(a=0.71),模擬計算的結果示于圖5。從圖5可以看出,隨著壓力的增大,長鏈烷烴脫氫轉化率和目標產物單烯烴的選擇性都呈下降趨勢。直鏈烷烴脫氫是可逆吸熱反應,壓力的增大降低了平衡轉化率;副反應多為不可逆反應,因而壓力的增大對于副反應的影響較小,因此在降低轉化率的同時,脫氫選擇性也呈下降趨勢。 3.2.3氫/烴摩爾比的影響 在絕熱脫氫反應器進口溫度480℃、絕對壓力0.24MPa、液時空速26h-1的條件下,改變氫/烴摩爾比,假定催化劑處于同一活性水平(a=0.71),模擬計算的結果示于圖6。從圖6可以看出,脫氫轉化率隨著氫/烴摩爾比的升高而降低,但目標產物單烯烴的選擇性隨氫/烴摩爾比的升高略有升高。在烴類原料進料量不變、反應壓力不變的情況下,增大氫/烴摩爾比降低了反應物的濃度,縮短了反應物料與催化劑的接觸時間,從而導致反應的轉化率下降。 3.3脫氫催化劑使用周期模擬結果 催化劑活性在操作周期的后期仍然處于較高水平,可考慮適當延長催化劑的使用周期。在催化劑實際使用55d時,直鏈烷烴脫氫轉化率為13.23%,選擇性為83.36%。通過計算發現,在本研究的工藝條件下,假如催化劑的使用周期延長7d,此時的直鏈烷烴脫氫轉化率為12.55%,選擇性為81.92%,與催化劑實際使用55d的轉化率相差0.68%,選擇性相差1.44%;假如將催化劑使用周期延長7d的反應器進口溫度提高2.5℃至480.5℃,反應壓力降低0.015MPa至0.21MPa,此時脫氫轉化率為13.12%,選擇性為81.96%,與催化劑實際使用55d相比,轉化率相差不多。由此可見在工業脫氫反應器中,催化劑的使用周期可以適當延長,有待工業上的進一步驗證。 4結論 (1) 采用一維擬均相絕熱反應器模型模擬長鏈烷烴工業脫氫反應器,結合長鏈烷烴的脫氫反應動力學模型,對工業脫氫反應器中直鏈烷烴脫氫反應進行了模擬計算。驗證結果表明,該模型可用于不同進口溫度、壓力以及氫/烴摩爾比的反應結果的定量預測。 (2) 在本研究的工藝條件下,確定了工業反應器催化劑失活動力學方程,催化劑的活性隨使用時間增長而下降,并成線性關系。失活動力學方程可用于催化劑活性變化的定量預測。 參考文獻 [1] 白子武, 趙玲. 直鏈烷基苯市場情況及發展預測[J].日用化學品科學,2006, 29(8): 1-3.(BAIZiwu,ZHAOLing.Marketsituationanddevelopmentoflinear-alkybenzene[J].Detergent&Cosmetics, 2006, 29(8):1-3) [2]LIRAA,TAILLEURRG.DehydrogenationofC12-C14paraffinsonPtCu/meso-structuredAl2O3catalystforLABproduction:Processsimulation[J].Fuel,2012, (97): 49-60. [3]IVASHKINAEN,YOURIEVEM,IVANCHINAED,etal.Developmentofanintelligentsystemforcontrollingparaffindehydrogenationcatalystoperationinproductionoflinearalkylbenzenes[J].EngineeringProblemsandIndustry, 2010, 2(2): 137-144. [4] 何松波, 孫承林, 杜鴻章,等. 長鏈烷烴(n-C10~13)脫氫制單烯烴催化劑的研究I. 氧化鋁載體孔結構對催化劑性能的影響[J].工業催化, 2009, 17(6): 46-49(HESongbo,SUNChenglin,DUHongzhang,etal.Investigationonthecatalystsfordehydrogenationoflongchainparaffins(n-C10~13)tomono-olefinsI.Effectofporestructureofaluminasupportonbehaviorsofthecatalyst[J].IndustrialCatalysis, 2009, 17(6): 46-49). [5]GALIASSOTR,DAVILAY.Optimalhydrogenproductionthroughrevampinganaphtha-reformingunit:Catalystdeactivation[J].EnergyFuel, 2008, 22(5):2892-2901. [6]FRANTSINAEV,IVASHKINAEN,IVANCHINAED,etal.Developingofthemathematicalmodelforcontrollingtheoperationofalkanedehydrogenationcatalystinproductionoflinearalkylbenzene[J].ChemicalEngineeringJournal, 2014, (238):129-139. [7] 江洪波,張晴, 周立群, 等. 長鏈烷烴脫氫制單烯烴動力學模型[J].石油學報(石油加工),2016,32(1):156-163.(JIANGHongbo,ZHANGQing,ZHOULiqun,etal.Kineticmodelofheavyparaffindehydrogenationtomono-olefins[J].ActaPetroleiSinica(PetroleumProcessingSection),2016,32(1):156-163.) [8] 朱炳辰, 翁惠新, 朱子彬. 催化反應工程(修訂版)[M].北京:中國石化出版社,2001. Kinetic Model of Heavy Paraffin Dehydrogenation to Mono-Olefins Ⅱ.Simulation of Industrial Dehydrogenation Reactor JIANG Hongbo1, WANG Yachun1, ZHANG Qing1, ZHOU Liqun2, WANG Yu2, CAO Jing2 (1. Research Institute of Petroleum Processing, East China University of Science and Technology, Shanghai 200237, China; 2. LAB Plant of Jinling Petrochemical Corporation of SINOPEC, Nanjing 210046, China) Keywords:heavyparaffin;dehydrogenation;radialreactor;deactivation;simulation Abstract:Withhigh-purityheavyparaffinasrawmaterial,thedehydrogenationofheavyparaffintomono-olefinswascarriedoutinanindustrialfixed-bedreactor,duringwhichthecatalystdeactivatedslowly.Basedonthematerialbalanceandheatbalanceequationsderivedfromone-dimensionalhomogenousadiabaticradialreactormodelanddehydrogenationkineticmodel,thedehydrogenationofheavyparaffinonindustrialreactorwassimulatedandtheinfluencesofoperatingconditionswerealsopredictedquantitatively.Thefittingresultofmodelwasgood,andthecatalystactivitydeclinedwithtimeshowingalinearrelationship.Theestablishedcatalystdeactivationequationcouldbeusedtoquantitativelypredictthechangeofcatalystactivityduringdehydrogenation. 收稿日期:2015-01-04 文章編號:1001-8719(2016)02-0388-06 中圖分類號:TQ032;TE65 文獻標識碼:A doi:10.3969/j.issn.1001-8719.2016.02.023 通訊聯系人: 江洪波,男,博士,副教授,研究方向石油加工動力學;E-mail:hbjiang@ecust.edu.cn