固相萃取-高效液相色譜法測定瀉痢消片中4種有效成分
2016-07-27 07:06:18吳江瑞齊廣才崔生飛張咪咪劉珍葉
西北藥學雜志 2016年4期
關鍵詞:固相萃取
吳江瑞,齊廣才,崔生飛,張咪咪,劉珍葉
(延安大學化學與化工學院,延安 716000)
?
固相萃取-高效液相色譜法測定瀉痢消片中4種有效成分
吳江瑞,齊廣才*,崔生飛,張咪咪,劉珍葉
(延安大學化學與化工學院,延安716000)
摘要:目的建立采用SPE-HPLC法同時測定瀉痢消片中芍藥苷、甘草苷、柚皮苷和橙皮苷含量的方法。方法采用C18固相萃取柱對瀉痢消片中的有效成分進行凈化、富集,用HPLC法測定含量。色譜柱為Kromasil C18柱(250 mm×4.6 mm,5 μm),流動相為乙腈-5 mL·L-1磷酸溶液,梯度洗脫,檢測波長為230,276和284 nm,柱溫為28 ℃,進樣量為20 μL,流速為1.0 mL·min-1。結果芍藥苷、甘草苷、柚皮苷和橙皮苷分別在0.617 0~30.85(r=1.000 0),0.159 8~7.944(r=0.999 8),0.972 8~48.64(r=1.000 0)和0.123 6~6.180 μg·mL-1(r=0.999 9)范圍內線性關系良好,平均回收率(n=6)分別為99.4%,98.3%,97.8% 和97.7%。RSD值分別為0.97%,1.11%,1.28% 和1.26%。結論該方法操作簡單快捷,靈敏度高,結果準確,可用于瀉痢消片中的4種有效成分的含量測定。
關鍵詞:瀉痢消片;芍藥苷;甘草苷;柚皮苷;橙皮苷;固相萃取;高效液相色譜法
瀉痢消片是由酒黃連、酒白芍、木香、枳殼、陳皮、茯苓、甘草等十二味中藥組方制成的治療腹瀉腹痛的中藥制劑;具有清熱燥濕、行氣鎮痛和化濁止痢的功效;主要用于大腸濕熱所致的腹痛泄瀉、大便不爽、里急后重、膿血便等癥[1-2]。方中酒白芍具有養血調經、柔肝止痛的作用,甘草的功能主要是補脾益氣、清熱解毒,并輔以枳殼和陳皮起到行滯消脹和理氣健脾的作用[3],對于治療腹瀉痢疾具有良好的效果。目前,關于瀉痢消片中有效成分含量的測定尚未見報道,為了更好地控制產品的質量,本文采用高效液相色譜法(HPLC)同時對其中芍藥苷[4]、甘草苷[5]、柚皮苷[6]和橙皮苷[7]4種有效成分進行含量測定,并運用固相萃取法(SPE)結合超聲提取對樣品進行前處理優化[8-13],以期多指標綜合評價其質量,所建立的SPE-HPLC分析方法能夠明顯減少雜質干擾,具有快速、簡捷、準確度高等優點,對瀉痢消片的質量控制有重要的指導意義。
1儀器與試藥
1.1儀器Agilent 1200型高效液相色譜儀
(含二極
管陣列檢測器);電子分析天平(AUY220型);超聲波清洗器(KQ-250B);固相萃取裝置(The BAKER spe-12G Glass Column Processor),Thermo HyperSep C18固相萃取柱(美瑞泰克科技公司)。
1.2試藥芍藥苷(批號110736-201438)、甘草苷(批號111610-201106)、柚皮苷(批號110722-200610)和橙皮苷(批號110721-201316)4種對照品均購自中國食品藥品檢定研究院;瀉痢消片(規格:每片0.35 g;批號:LMA1304,LMA1309,LEA1409;云南白藥集團麗江藥業有限公司);甲醇、乙腈均為色譜純(美國DIKMA公司);水為超純水;其余試劑均為分析純。
2方法與結果
2.1色譜條件色譜柱:Kromasil C18柱(250 mm×4.6 mm,5 μm)。流動相:乙腈(A)-5 mL·L-1磷酸溶液(B),梯度洗脫(0~6 min,24% A;6~10 min,24% A→30% A)。流速:1.0 mL·min-1;波長切換:0~6 min,230 nm;6~7 min,276 nm;7~10 min,283 nm。柱溫:28 ℃,進樣量:20 μL,峰面積外標法定量。色譜圖見圖1。
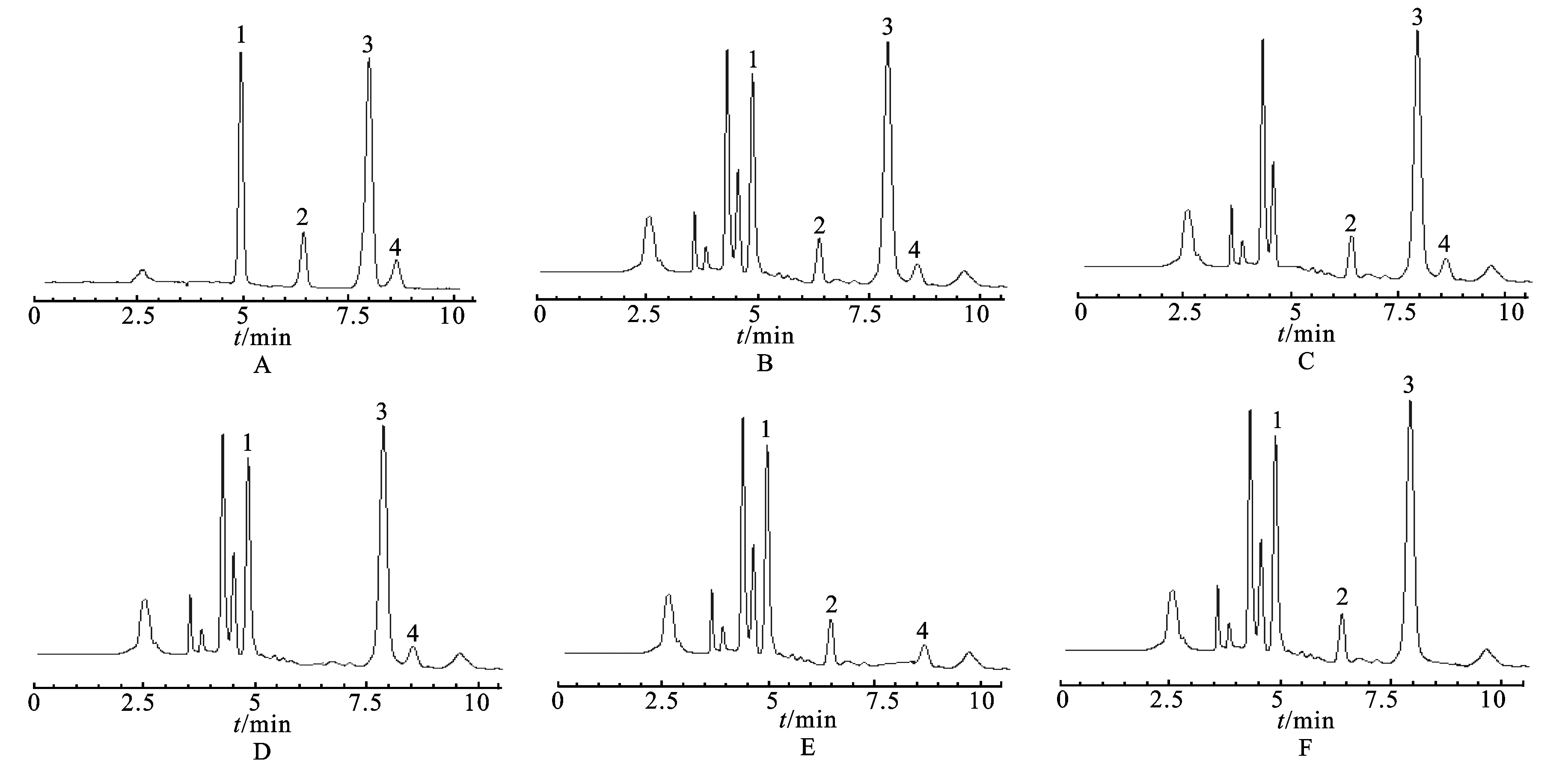
圖1瀉痢消片HPLC圖譜
A.對照品;B.樣品;C.缺白芍陰性樣品;D.缺甘草陰性對照;E.缺枳殼陰性對照;F.缺陳皮陰性對照;1.芍藥苷;2.甘草苷;3.柚皮苷;4.橙皮苷
Fig.1 HPLC chromatograms of Xielixiao Tablets
A.reference substances;B.sample;C.negative sample without RadixPaeoniaeAlba; D.negative sample withoutLicorice;E.negative sample withoutFructusAurantii;F.negative sample withoutCitrus;1.paeoniflorin; 2.liquiritin;3.naringin; 4.hesperidin
2.2溶液的制備
2.2.1對照品溶液的制備準確稱取對照品芍藥苷、甘草苷、柚皮苷和橙皮苷適量,分別置于25 mL量瓶中,用甲醇溶解并稀釋至刻度,搖勻,制得質量濃度分別為192.8,198.6,152.0和308.8 μg·mL-1的單一成分對照品儲備液。分別精密量取芍藥苷對照品儲備液4.0 mL、甘草苷對照品儲備液1.0 mL、柚皮苷對照品儲備液8.0 mL和橙皮苷對照品儲備液0.5 mL,置于同一25 mL量瓶中,加甲醇稀釋至刻度,搖勻,即得混合對照品儲備液Ⅰ(芍藥苷30.85 μg·mL-1,甘草苷7.944 μg·mL-1,柚皮苷48.64 μg·mL-1,橙皮苷6.180 μg·mL-1)。
2.2.2供試品溶液的制備取瀉痢消片適量,除去包衣,置于研缽,研細混勻,取其粉末約1.0 g,精密稱定,置于100 mL具塞三角瓶中,精密加甲醇25.00 mL,密塞,稱定質量,超聲提取20 min,放冷,再稱定質量,用甲醇補足減失的質量,搖勻,用0.45 μm微孔濾膜濾過,精密量取所得續濾液10 mL上樣至預先用甲醇(5 mL×2)活化,水(5 mL×2)平衡的固相萃取柱中,然后用5 mL體積分數為3%的甲醇淋洗樣品,以小于2 mL·min-1的流速淋洗,棄去全部流出液;用5 mL體積分數為80%的甲醇洗脫,收集洗脫液至10 mL量瓶中。
2.2.3陰性樣品溶液的制備按照處方比例及制備工藝,取處方中除白芍、甘草、枳殼和陳皮外的其他藥味,制得陰性對照樣品,按照2.2.2項下方法制成陰性對照溶液。
2.3方法學考察
2.3.1線性關系考察按照2.1項下色譜條件,分別精密吸取混合對照品儲備液Ⅰ0.50,1.00,3.00,5.00,10.00和25.00 mL,分別置于25 mL量瓶中,用甲醇稀釋至刻度,搖勻,制成系列質量濃度對照品混合溶液。分別精密吸取上述系列對照品混合溶液各20 μL注入色譜儀,以色譜峰面積Y對對照品質量濃度X(μg·L-1)進行線性擬合。求得芍藥苷、甘草苷、柚皮苷和橙皮苷回歸方程分別為:Y1=15.626X1-0.891 2(r1=1.000 0),Y2=18.185X2+5.325 2(r2=0.999 8),Y3=16.057X3+0.239 2(r3=1.000 0),Y4=17.492X4+2.096 8(r4=0.999 9)。結果表明,芍藥苷、甘草苷、柚皮苷和橙皮苷分別在0.617 0~30.85,0.159 8~7.944,0.972 8~48.64和0.123 6~6.180μg·mL-1范圍內線性關系良好。
2.3.2精密度實驗按照2.1項下色譜條件,精密吸取對照品混合儲備液Ⅰ20μL,重復進樣6次,測得芍藥苷、甘草苷、柚皮苷和橙皮苷等峰面積的RSD值分別為0.61%,0.86%,1.08%和2.34%。儀器精密度符合要求。
2.3.3重復性實驗按2.2.2項下方法平行制備供試品溶液6份,取同一批號(批號LMA1304)樣品,在上述色譜條件下分別進樣20μL,測得峰面積,結果芍藥苷、甘草苷、柚皮苷和橙皮苷的RSD分別為1.31%,2.68%,0.92%和2.61%,實驗結果表明重復性良好。
2.3.4穩定性實驗精密吸取同一供試品溶液20μL,按照上述色譜條件,分別在0,2,4,6,8和10h進樣,測得芍藥苷、甘草苷、柚皮苷和橙皮苷峰面積的RSD值分別為0.82%,1.27%,0.32% 和1.18%。結果表明,供試品溶液在10h內穩定。
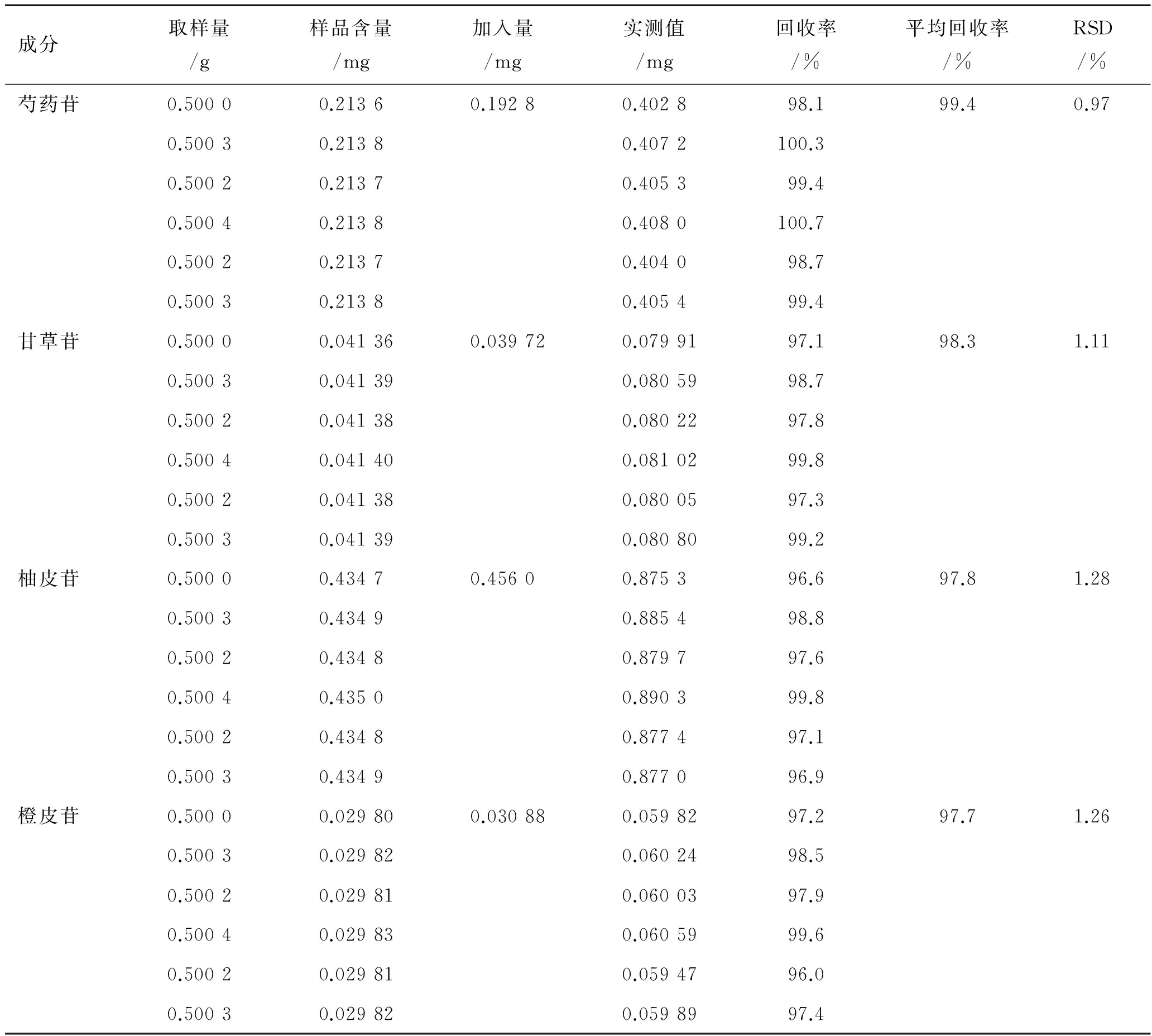
2.3.5加樣回收率實驗精密稱取已知含量的樣品(批號LMA1304)6份,每份約0.5g,分別精密加入芍藥苷對照品溶液(質量濃度為192.8μg·mL-1)1.0mL,甘草苷對照品溶液(質量濃度為198.6μg·mL-1)0.2 mL,柚皮苷對照品溶液(質量濃度為152.0 μg·mL-1)3.0 mL,橙皮苷對照品溶液(質量濃度為308.8 μg·mL-1)0.1 mL,按照2.2.2項下方法制備供試品溶液,測定含量并計算回收率,結果見表1。
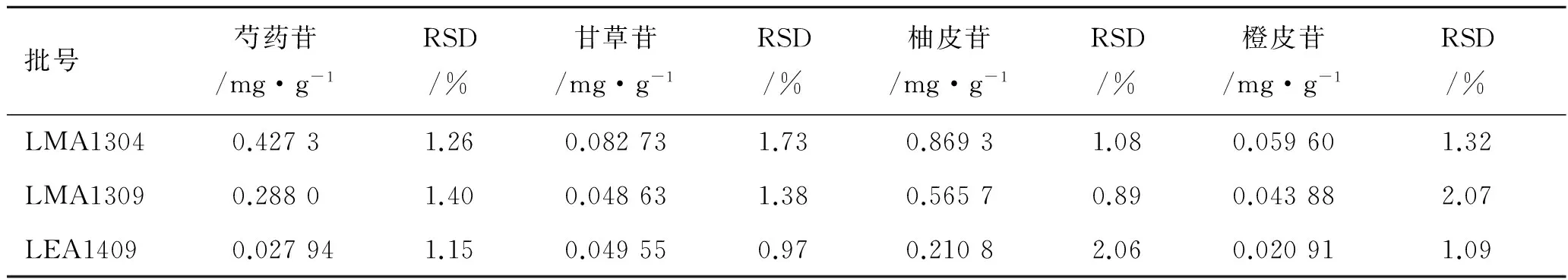
2.4樣品含量測定取3批樣品,每批樣品取5份,按照2.2.2項下方法分別制備供試品溶液,按照2.1項下色譜條件進樣,測定峰面積,按照外標法計算含量,結果見表2。
3討論
3.1提取方法的選擇(1)實驗用水、甲醇、乙醇和乙腈4種不同溶劑提取樣品,結果表明,用甲醇對樣品進行提取優于其他3種溶劑,且與雜質能夠有效分離。(2)采用超聲法、固相萃取法、超聲法結合固相萃取法對樣品進行處理,比較各方法可知,超聲法提取后樣品中的雜質干擾較嚴重;直接用固相萃取法不能將樣品完全提取;超聲提取后,再進行固相萃取雜質干擾少,對樣品成分的提取效率高,故選用此法。(3)比較超聲處理時間10,20,40,60,80和100 min,發現超聲20 min即能將被測成分提取完全。(4)比較用甲醇溶液5,15,25,40,50和60 mL提取樣品,結果用25 mL甲醇溶液提取的含量最高。
表1加樣回收率實驗結果
Tab.1 Results of recovery tests
(n=6)
表2樣品含量測定結果
Tab.2 Determination results of samples
(n=5)
3.2固相萃取條件的優化
3.2.1固相萃取柱的選擇實驗中比較了Thermo HyperSep C18固相萃取柱和Thermo Hy- perSep Retain PEP固相萃取小柱的萃取效果,結果發現二者對芍藥苷和橙皮苷的提取相差甚小,而C18固相萃取柱對甘草苷和柚皮苷的提取優于PEP固相萃取小柱,且分離徹底,故選用Thermo HyperSep C18固相萃取柱。
3.2.2淋洗溶劑的優化分別取樣品0.5,2.0,4.0,6.0,8.0和10.0 mL對上樣體積進行考察,由于樣品中雜質干擾較嚴重,當上樣體積大于8.0 mL時,顯示出較好的抗干擾效果,為保證回收率,上樣體積選取10.0 mL。用水,體積分數分別為1%,3%,5%,8%和10%的甲醇為淋洗溶劑,收集淋洗液,用HPLC檢測,結果顯示,用體積分數為3%的甲醇淋洗時任何目標物不被洗脫,為了減少雜質的干擾并得到最佳的提取效果,確定用體積分數為3%的甲醇淋洗溶液。
3.2.3洗脫條件的優化選擇不同體積分數的甲醇(5%,25%,45%,65%,80% 和100%)溶液進行洗脫,用體積分數為80%的甲醇洗脫,雜質干擾少,提取率也較高。選取洗脫體積(1,3,5,7,9和10 mL)時,用5和7 mL結果并無差異,為了洗脫完全又不浪費溶劑,最終選取5 mL進行洗脫。
3.3檢測波長的選擇樣品中4種苷類物質的色譜峰經二極管陣列檢測器檢測,其UV光譜與各自對照品的UV光譜一致,芍藥苷在230 nm處有最大吸收峰,甘草苷在276 nm處有最大吸收峰,柚皮苷和橙皮苷均在283 nm處有最大吸收峰,因此均選用各自最大吸收峰處的波長作為實驗測定波長。
3.4流動相的選擇比較流動相(A)甲醇-水,(B)乙腈-2 mL·L-1磷酸水溶液,(C)乙腈-5 mL·L-1磷酸水溶液,結果顯示,3種流動相均可達到分離效果,但是用流動相(A)雜質峰干擾嚴重,而在流動相中加入磷酸(B、C)抑制苷中酚羥基的離解,實驗重復性好。(C)比(B)使柚皮苷和橙皮苷的峰形更好,最終選取(C)作為實驗的流動相。
參考文獻:
[1]李宜航,路娟,李光,等.瀉痢消片治療實驗性結腸炎的研究[J].醫藥導報,2013,32(7):881-884.
[2]李世輝,朱虹江.瀉痢消膠囊治療濕熱泄瀉108例臨床觀察[J].云南中醫學院學報,2009,32(3):41-44.
[3]國家藥典委員會.中國藥典2010年版[S].一部.北京:中國醫藥科技出版社,2010:81-230.
[4]劉阿靜,齊廣才,劉珍葉,等.RP-HPLC法測定防風通圣丸中芍藥苷、梔子苷、黃芩苷和連翹苷的含量[J].西北藥學雜志,2012,27(5):415-417,437.
[5]霍生青,張耀元,張志成,等.HPLC單波長同時測定六味甘草丸中的甘草苷和甘草酸銨[J].華西藥學雜志,2014,29(2):199-200.
[6]牛曉靜,魯靜,段曉穎,等.HPLC同時測定健脾舒胃凝膠中甘草苷、柚皮苷、橙皮苷、新橙皮苷、甘草酸銨5種成分含量[J].中國實驗方劑學雜志,2015,21(2):77-79.
[7]侯振山,蔣志濤,毛敏玨,等.HPLC法測定肝爾舒中柚皮苷和橙皮苷的含量[J].西北藥學雜志,2012,27(4):305-307.
[8]張赟華,董媛,李忠瓊,等.HPLC-DAD法同時測定氣滯胃痛顆粒中5個成分的含量[J].藥物分析雜志,2012,32(9):1661-1664.
[9]李文博,韓建平,倪倩,等.SPE-UPLC法測定養血清腦顆粒中的芍藥苷含量[J].藥物分析雜志,2011,31(7):1385-1388.
[10]黃莉,夏新華,譚喜平.SPE-HPLC法同時測定荊防小兒止咳口服液中升麻素苷和連翹苷[J].中成藥,2014,36(6):1203-1208.
[11]張西如,趙江麗,姜建國,等.SPE-HPLC測定小兒氨酚黃那敏顆粒中4種合成色素[J].中國現代應用藥學,2013,30(10):1113-1116.
[12]丘明明,黃玉華.SPE-HPLC法測定胃腸寧片中3種生物堿的含量[J].中成藥,2011,33(2):276-279.
[13]郭懷忠,張斌,王配,等.SPE-HPLC法測定藿香正氣水中厚樸酚與和厚樸酚的含量[J].藥物分析雜志,2010,30(5):827-830.
基金項目:延安大學研究生教育創新計劃項目
作者簡介:吳江瑞,女,碩士研究生
*通信作者:齊廣才,男,教授,碩士研究生導師
doi:10.3969/j.issn.1004-2407.2016.04.016
中圖分類號:R927.2
文獻標志碼:A
文章編號:1004-2407(2016)04-0379-05
(收稿日期:2015-09-15)
Determination of 4 kinds of effective components in Xielixiao Tablets by SPE-HPLC
WU Jiangrui,QI Guangcai*,CUI Shengfei,ZHANG Mimi,LIU Zhenye
(College of Chemistry and Chemical Engineering,Yan′an University,Yan′an 716000,China)
Abstract:Objective To establish a solid-phase extraction with high-performance liquid chromatography(SPE-HPLC) method for the determination of paeoniflorin,liquiritin,naringin,and hesperidin in Xielixiao Tablets.Methods Using C18solid phase extraction column,the active ingredients of Xielixiao Tablets were purified and enriched.The contents were analyzed by HPLC method with Kromasil C18column(250 mm×4.6 mm,5 μm).The mobile phase was composed of acetonitrile and 5 mL·L-1phosphoric acid in gradient elution.Detection wavelength was set at 230,276 and 284 nm.The column temperature was maintained at 28 ℃,and injection volume was 20 μL.The flow rate was 1.0 mL·min-1.Results The calibration curves of paeoniflorin,liquiritin,naringin,and hesperidin showed good linearity within 0.617 0-30.85(r=1.000 0),0.159 8-7.944(r=0.999 8),0.972 8-48.64(r=1.000 0),and 0.123 6-6.180 μg·mL-1(r=0.999 9),respectively.The average recoveries were 99.4%,98.3%,97.8% and 97.7%.RSDs were 0.97%,1.11%,1.28% and 1.26%,respectively.Conclusion The method is simple,fast,sensitive,and accurate,and can be used for the quality control of 4 kinds of effective components in Xielixiao Tablets.
Key words:Xielixiao Tablets;paeoniflorin;liquiritin;naringin;hesperidin;SPE;HPLC
猜你喜歡
分析化學(2016年7期)2016-12-08 00:54:07
中國科技博覽(2016年2期)2016-04-25 14:11:43
湖北工業職業技術學院學報(2016年1期)2016-04-20 17:12:54
分析化學(2015年10期)2015-11-03 07:52:24
食品安全導刊(2015年10期)2015-10-26 04:44:22
安徽農學通報(2015年18期)2015-10-20 00:50:11
安徽農學通報(2015年17期)2015-09-30 00:52:24
分析化學(2015年9期)2015-09-11 07:09:54
肉類研究(2015年5期)2015-08-08 12:46:08
肉類研究(2015年3期)2015-06-16 12:40:36
