Lactobacilluskefiranofaciens乳糖酶基因克隆及在畢赤酵母中表達
2016-09-18 12:38:21邢竹青王彥寧劉兆賢鄔亞男王艷萍天津科技大學食品工程與生物技術學院天津300457
中國釀造 2016年1期
邢竹青,王彥寧,劉兆賢,鄔亞男,梁 麗,王艷萍(天津科技大學 食品工程與生物技術學院,天津 300457)
Lactobacilluskefiranofaciens乳糖酶基因克隆及在畢赤酵母中表達
邢竹青,王彥寧,劉兆賢,鄔亞男,梁麗,王艷萍*
(天津科技大學 食品工程與生物技術學院,天津 300457)
以馬乳酒樣乳桿菌ZW3基因組為模板,PCR擴增得到乳糖酶中LacL、LacM兩個大小亞基片段,經EcoRI和SnaBI雙酶切后,分別連接pPIC9K質粒,轉化大腸桿菌感受態細胞DH5α,并驗證其核苷酸序列正確。將重組質粒pPIC9K-LacL、pPIC9K-LacM分別電轉化畢赤酵母GS115,構建pPIC9K-LacL-GS115重組子和pPIC9K-LacM-GS115重組子,通過篩選得到抗G418濃度達到3.0mg/m L,具有多拷貝基因的重組子。經反轉錄PCR驗證,cDNA上存在LacL片段,并可檢測到乳糖酶酶活達到1.17 U/m L;pPIC9K-LacM-GS115重組子誘導表達72 h后的發酵液上清經SDS蛋白電泳檢測到目的條帶。
乳糖酶;畢赤酵母;克隆;表達
乳糖酶水解乳糖的糖苷鍵分解乳糖為半乳糖和葡萄糖,或發生轉糖苷作用生成低聚半乳糖[1]。主要用于生產低乳糖牛奶以緩解乳糖不耐受癥,近年來其在保健食品生產領域的應用也備受關注(如低聚半乳糖[2]),其市場需求量逐年擴大。因此,對乳糖酶的研究不僅具有理論意義,而且有巨大的社會價值和經濟效益。
乳酸菌被公認為一般安全(generally recognizedassafe,GARS)食品,是應用于食品工業中的重要菌種[3]。乳酸菌來源的乳糖酶為中性乳糖酶[4],其最適溫度介于40~60℃,可滿足乳制品生產的要求,但其不能胞外分泌,提高了分離純化的難度,不適宜實際食品工業生產。為了獲得具有大規模工業生產價值的重組酶發酵菌株,一株經過全基因組測序的馬乳酒樣乳桿菌ZW 3[5]中兩個含有重疊區域的LacL、LacM乳糖酶大小亞基片段分別被克隆到高效的蛋白表達平臺——畢赤酵母[6]中,進行了初步表達。
1 材料與方法
1.1料與試劑
1.1.1種
馬乳酒樣乳桿菌(Lactobacilluskefiranofaciens)ZW 3,大腸桿菌DH5α,畢赤酵母(Pichia pastoris)GS115,以上菌種均為本實驗室保藏。
1.1.2養基[6-7]
LB培養基、MRS培養基、酵母浸出粉胨葡萄糖(yeast extract peptone dextrose,YPD)培養基、MD培養基、基本培養基(m inimalmedium,MM)、BMGY誘導表達培養基、BMMY誘導表達培養基。
1.1.3粒
酵母分泌型表達載體pPIC9K:由本實驗室保存。
1.1.4物
基因Lac L和LacM引物:根據大小亞基基因片段的核酸序列設計引物,上游引物添加限制性內酶位點SnaB I,下游引物添加限制性內切酶位點EcoR I。
Lac L F 5′-CGGTACGTAATGCAAGCAAATATTAA ATG-3′
Lac L R 5′-CGCGGGAATTCTTATTTGTGTAATCC ATAATAG-3′
Lac M F 5′-CCGTACGTAATGGATTACACAAATA AG-3′
LacM R 5′-CGGAATTCTTAAAACTGGTTTAAGAT GAAGG-3′
pPIC9K載體測序通用引物:
5′AOX1 5′-GACTGGTTCCAATTGACAAGC-3′
3′AOX1 5′-GCAAATGGCATTCTGACATCC-3′
1.1.5要試劑
限制性內切酶SnaB I及EcoR I、T4連接酶、Fastpfu高保真聚合酶、細菌基因組提取試劑盒、質粒提取試劑盒、PCR純化試劑盒、反轉錄試劑盒、β-半乳糖苷酶試劑盒。
1.2器與設備
genepulser Xcell電轉儀、聚合酶鏈式反應(polymerase chain reaction,PCR)儀、Power PAC 3000電泳儀、全自動凝膠成像儀:美國Bio-Rad公司;DYY-Ⅲ-6B型穩壓穩流電泳儀、DYCZ-24D型電泳槽:北京六一儀器廠;5415D型離心機:德國Eppendorf公司;HZQ-F160全溫振蕩培養箱:北京東聯哈爾儀器制造有限公司;AD07R-20-A12E循環水浴鍋:美國Polyscience公司;VIS-7220型分光光度計:北京瑞利分析儀器公司。
1.3法
1.3.1色體的提取
采用細菌基因組提取試劑盒提取馬乳酒樣乳桿菌ZW 3染色體。
1.3.2糖酶大、小亞基基因片段的擴增
以馬乳酒樣乳桿菌ZW 3染色體為模板、LacL F、LacL R、LacM F、LacM R為引物,以常規方法擴增,擴增參數:94℃預變性4min;94℃變性30 s,55℃退火30 s,72℃延伸2m in,共進行30個循環;最后72℃延伸10m in。
1.3.3別構建pPIC9K-LacL、pPIC9K-LacM重組質粒
擴增所得的大、小亞基片段進行SnaB I和EcoR I雙酶切,分別與經過相應限制性內切酶雙酶切后的pPIC9K質粒,在T4連接酶的作用下,16℃進行連接過夜。連接產物轉化大腸感受態細胞DH5α,驗證陽性的克隆,送華大生物進行測序驗證。
1.3.4Pichia pastorisGS115感受態制備
參照參考文獻[7]。
1.3.5化Pichia pastoris GS115
參照《分子克隆指南》酵母電轉化方法[8]。
1.3.6赤酵母重組子總RNA提取與反轉錄
Trizol法[9]提取畢赤酵母重組子總RNA,以提取到的總RNA作為模板利用反轉錄試劑盒,合成cDNA。
1.3.7糖酶酶活力的測定
使用β-半乳糖苷酶試劑盒測定,按照說明書操作。單位的定義:每1m L發酵上清液每小時產生1 nmol對硝基苯酚定義為一個酶活力單位,U/m L。
2 結果與分析
2.1PCR擴增馬乳酒樣乳桿菌乳糖酶基因
在NCBI中查找馬乳酒樣乳桿菌ZW 3全基因組序列上乳糖酶基因LacLM,全長2 833 bp,由大小兩個亞基構成,其中大亞基LacL全長1 884 bp,小亞基LacL全長960 bp,二者有重疊區域。利用BLAST軟件進行蛋白相似性比對,可知與LacL蛋白相似性最高的序列為瑞士乳桿菌中的乳糖酶大亞基,相似度達到82%;與LacM蛋白相似性最高的序列為瑞士乳桿菌中的乳糖酶小亞基,相似度達到81%。因為畢赤酵母表達載體pPIC9K已經帶有信號肽序列,所以不需要添加信號肽。PCR擴增結果(圖1)所示,產物分別在不到1 000 bp處和接近2 000 bp處有兩條單一條帶,圖1中顯示的結果與理論大小一致。
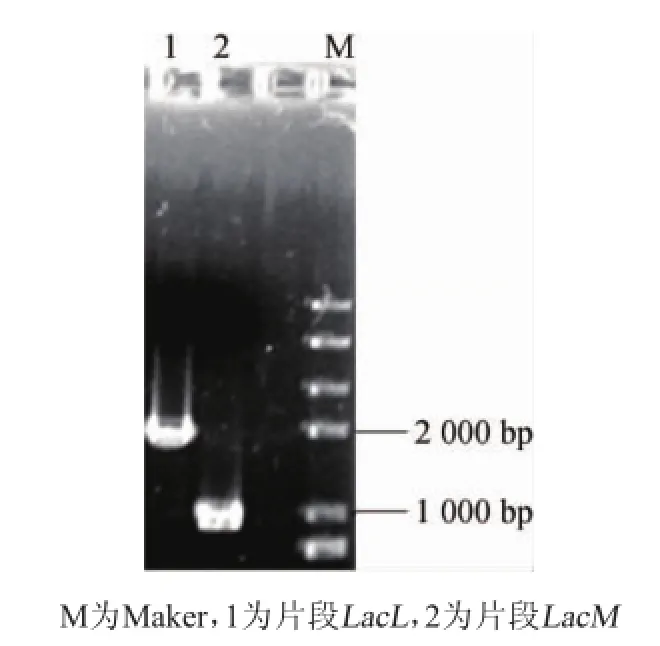
圖1 乳糖酶基因LacL、LacM PCR擴增產物電泳結果Fig.1 Electrophoretogram of PCR amp lification products of lactase gene LacL and LacM
2.2組載體的驗證
提取重組子中質粒,進行SnaB I和EcoR I雙酶切驗證,重組載體斷為兩條帶。結果如圖2所示。
由圖2可知,大于8 000 bp的條帶為pPIC9K載體片段,接近2 000 bp的片段為LacL片段,接近1 000 bp的片段為LacM片段,與目標條帶大小一致。重組子質粒經過通用引物5’AOX1和3’AOX1測序后,與馬乳酒樣乳桿菌ZW 3基因組上大、小亞基片段的核苷酸序列進行比對,結果相似性為100%,說明所得大、小亞基的核苷酸序列完全正確。
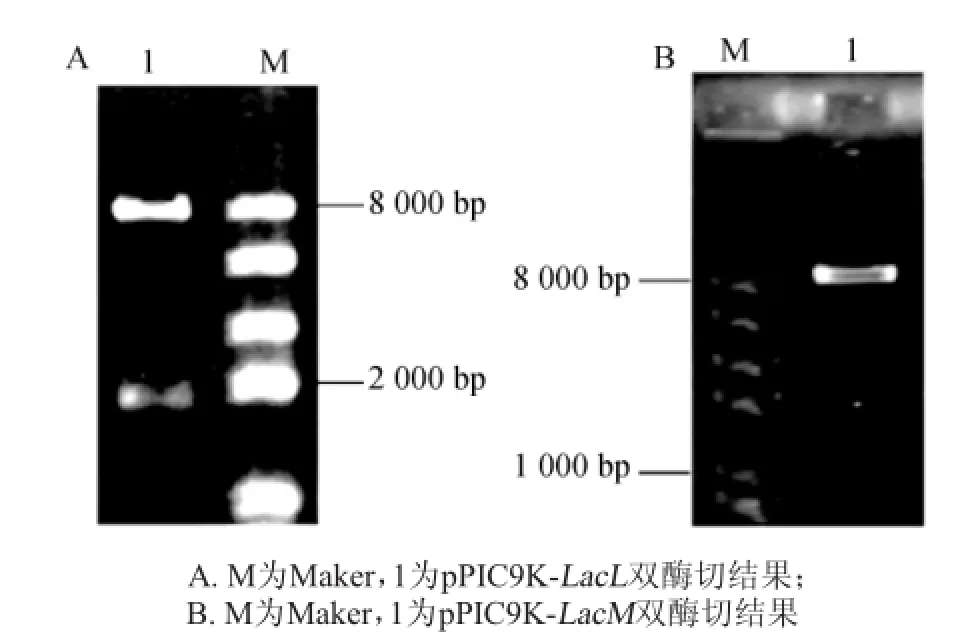
圖2 重組pPIC9K-LacL質粒(A)和重組pPIC9K-LacM質粒(B)驗證結果Fig.2 Verification results of recom binant p lasm id pPIC9K-LacL(A)and pPIC9K-LacM(B)
2.3組質粒pPIC9K-LacL、pPIC9K-LacM電轉化Pichia pastoris GS115
重組質粒pPIC9K-LacL、pPIC9K-LacM分別電擊轉化感受態細胞Pichiapastoris GS115。轉化子經MM/MD平板篩選后,挑選His+Mut+表型的酵母重組子,利用通用引物5’AOX1和3’AOX1進行驗證。由圖3(A)可知,1-7泳道均出現兩條帶,一條為AOX 1基因的2.2 kbp條帶,另一條為目的條帶大小,接近2 000 bp,與LacL片段大小一致。圖3(B)中,2和3泳道只有目的基因一條帶,說明AOX1基因在整合過程中被破壞,該重組子為假陽性;另外4個泳道,均出現兩條帶,一條為AOX1基因的2.2 kbp條帶,另一條為目的條帶,大小接近1 000 bp,與LacM片段大小一致。從而證明,LacL與LacM基因已分別成功整合到Pichia pastoris GS115基因組中。
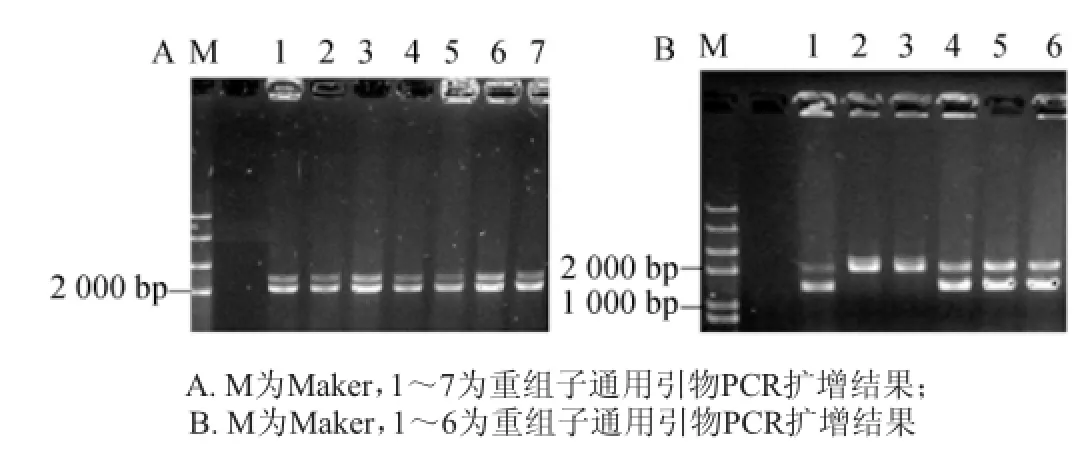
圖3 pPIC9K-Lac L酵母重組子(A)和pPIC9K-LacM酵母重組子(B)PCR驗證結果Fig.3 PCR verification resu lts of yeast recombinant pPIC9K-LacL(A)and pPIC9K-LacM(B)
2.4拷貝重組子的篩選
pPIC9K質粒整合在酵母染色體中,能夠使細胞產生G418抗性,其G418抗性水平與整合的卡那霉素抗性基因拷貝數直接相關,可進行體內多拷貝整合的篩選。分別取菌體細胞依次點接于含不同質量濃度(0.50mg/m L、1.00mg/m L、2.00mg/m L、3.00 mg/m L)G418抗性平板上。以pPIC9KLacL-GS115酵母重組子為例,30℃培養2~5 d,挑取4株在質量濃度為3.00mg/m L的G418抗性平板上生長較好的菌落,作為乳糖酶誘導表達的待選菌株,同樣方法選取4株pPIC9K-LacM-GS115酵母重組子,作為待選菌株。
2.5亞基LacL與小亞基LacM分別在畢赤酵母GS115中的表達
將上述待選菌株接種于含有G418抗性的25m L BMGY培養基,30℃振蕩培養至對數生長期,以轉化pPIC9K空載體的畢赤酵母GS115菌株作為對照。再轉接于適當體積的BMMY培養基中至OD600值為1.0~2.0,30℃振蕩培養,開始誘導表達,每隔24 h補加甲醇使其最終體積分數為0.5%以維持誘導。將選取的pPIC9K-LacL-GS115重組子和pPIC9K-LacM-GS115重組子各4株,經甲醇誘導表達后,取培養72 h后的發酵液上清液進行SDS-PAGE分析。如圖4所示,4株pPIC9K-LacM-GS115重組子在約40 ku左右均有1條特異蛋白條帶,與預期得到帶有信號肽的目標蛋白分子量相一致;在>40 ku處也出現一條特異性條帶,通過NetNGlyc 1.0服務器分析,可知乳糖酶LacM氨基酸序列有一個潛在的N-糖基化位點,由此推測由于部分蛋白經過糖基化修飾,導致分子量偏大。而在圖4中pPIC9K-LacL-GS115 4株重組子在80 ku處無明顯可見特異性蛋白條帶,這可能是由于表達量偏低,條帶不易檢測。
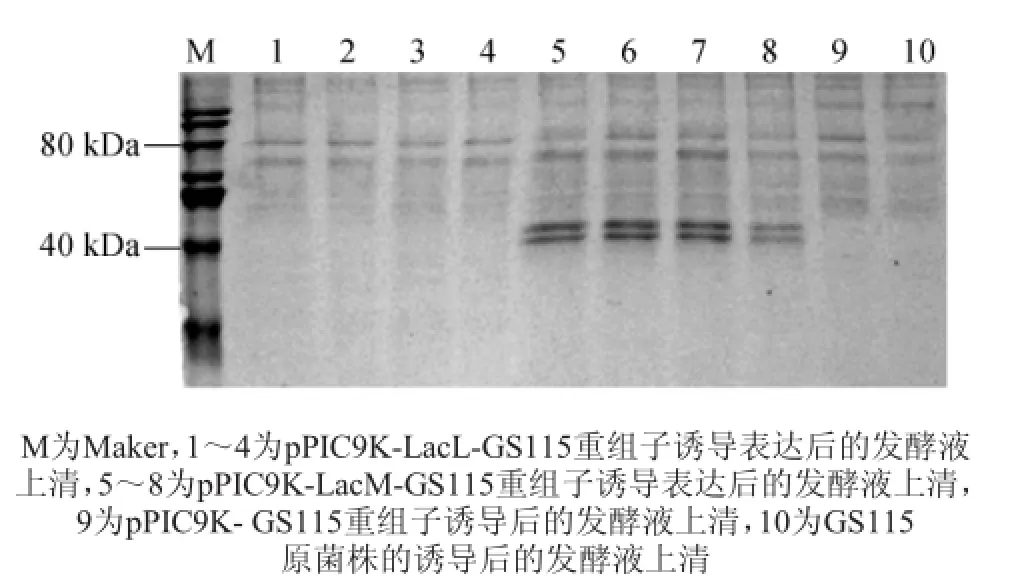
圖4 重組子誘導表達蛋白電泳圖Fig.4 Electrophoretogram of inducible expression protein by recombinant
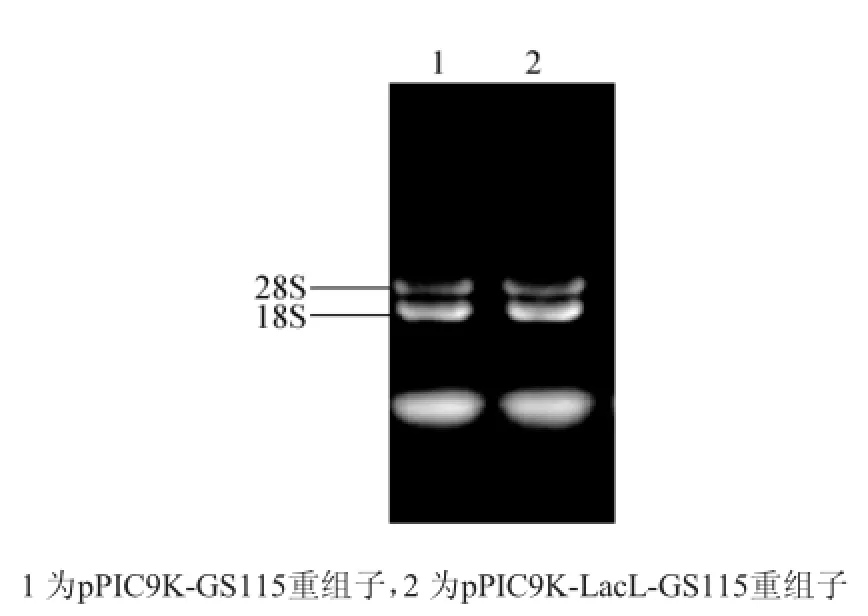
圖5 酵母重組子RNA提取結果Fig.5 RNA extraction resultof yeast recombinant
為了檢測pPIC9K-LacL-GS115重組子中的LacL片段是否進入到轉錄階段,提取重組子總RNA(如圖5),通過反轉錄得到相應的cDNA作為模板,以LacL片段的特異引物進行PCR,以驗證插入的LacL片段是否有相應的mRNA轉錄(如圖6)。由圖5可知,LacL片段在pPIC9K-LacL-GS115重組子中進行了轉錄過程,經過稀有密碼子軟件[10]分析發現編碼LacL大亞基片段的基因序列在畢赤酵母中翻譯時有12個氨基酸的密碼子使用頻率低于10%,故推測是翻譯過程效率較低,導致蛋白表達量低。
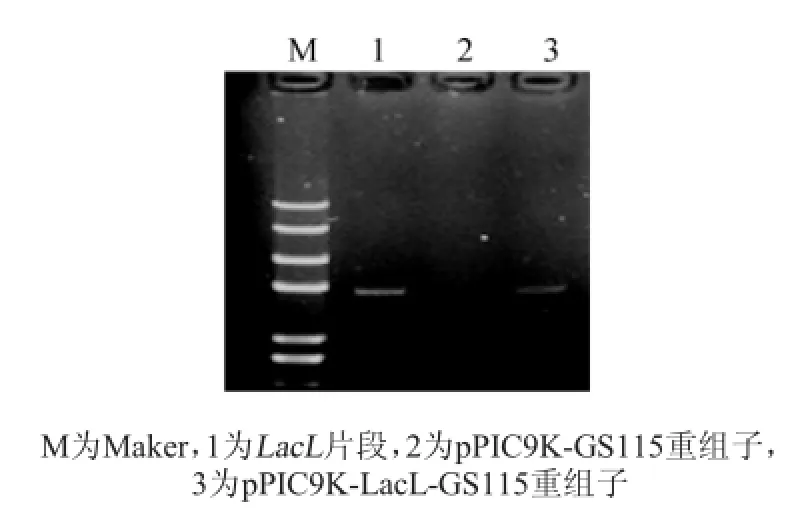
圖6 cDNA PCR驗證結果Fig.6 cDNA PCR identification result
現有研究顯示不同來源的乳糖酶基因在相似條件下經甲醇誘導在畢赤酵母中表達,重組酶酶活水平在0.37~51.2U/m L[11-12],本實驗中pPIC9K-LacL-GS115重組子誘導表達72 h后的發酵液上清乳糖酶酶活達到1.17 U/m L,陰性對照組pPIC9K-GS115重組子甲醇誘導后未能檢測到乳糖酶活力,而pPIC9K-LacM-GS115重組子發酵72 h后的上清液未能測出乳糖酶酶活,這可能是由于其在畢赤酵母中糖基化后改變構象造成的。另外,據文獻報道[13-15],LacL和LacM二者多以異源二聚體的形式發揮較高的乳糖酶活性。
3 結論
目前工業化生產使用的乳糖酶多來源于克魯維酵母,生產過程中必須經過破壁提取酶蛋白,造成乳糖酶收率低,影響酶活力[16]。利用畢赤酵母表達系統進行外源分泌表達,可使酶直接分泌于胞外,簡化工藝步驟,減少酶活損失。本文采用畢赤酵母表達系統進行重組乳糖酶的表達,成功將馬乳酒樣乳桿菌ZW 3中乳糖酶大小亞基進行克隆,并分別連接到高效表達載體pPIC9K上,整合至畢赤酵母GS115中。重組菌株對G418的抗性越強表明外源基因整合的拷貝數越高[17],實驗中成功篩選出高拷貝的pPIC9K-LacLGS115重組子和pPIC9K-LacM-GS115重組子。pPIC9KLacM-GS115重組子誘導表達72 h后的發酵上清液中檢測到目的蛋白LacM相應條帶,并且測定出pPIC9K-LacLGS115重組子誘導表達72 h后的發酵上清液中乳糖酶酶活達到1.17U/m L。本研究在畢赤酵母中成功表達了來自馬乳酒樣乳桿菌ZW 3中的乳糖酶大小亞基,為其應用研究奠定了基礎。
[1]黃慧福.乳糖酶的性質及其應用[J].農技服務,2010,27(10):1353-1354.
[2]張群.低聚半乳糖生產技術研究[J].食品與生物技術學報,2015,34(7):784-784.
[3]閆肅,呂嘉櫪,郜洪濤.乳酸菌在食品工業中應用[J].中國釀造,2010,29(12):1-3.
[4]章沙沙,張宇宏,樊曉虎,等.乳桿菌乳糖酶的基因異源表達及酶學性質分析[J].中囯農業科技導報,2011,13(3):53-59.
[5]WANG Y P,WANG JR,ZAHEER A.Complete genome sequence of Lactobacillus kefiranofaciens ZW 3[J].J Bacteriol,2011,193(16):4280-4281.
[6]顧小勇,李強.畢赤酵母基因工程菌胞內AOX酶的檢測方法[J].生物工程學報,2001,17(4):474-477.
[7]王慧.應用雙質粒體系提高融合蛋白GGH在畢赤酵母GS115中表達量的初步研究[D].無錫:江南大學碩士論文,2011.
[8]J.薩姆布魯克,E.F.弗里奇,T曼尼阿蒂斯.分子克隆實驗指南[M].北京:科學出版社,1992.
[9]于寒松,彭帥,謝遠紅,等.一種RNA提取試劑盒——TRIZOL的使用方法初探[J].食品科學,2005,26(11):39-42.
[10]FUHRMANN M,HAUSHERR A,FERBITZ L,et al.M onitoring dynamic expression ofnucleargenes in Chlamydomonasreinhardtii by using a synthetic luciferase reportergene[J].Plant M ol Biol,2004,55(6): 869-881.
[11]王霽昀.耐熱β-半乳糖苷酶的克隆表達研究[D].無錫:江南大學碩士論文,2009.
[12]聶春明.乳酸桿菌β-半乳糖苷酶重疊基因的克隆表達及酶學性質分析[D].呼和浩特:內蒙古農業大學碩士論文,2012.
[13]SANTORO L,CAMMAROTA G,MANNA R,et al.M anagement and treatment of lactosemalabsorption[J].World J Gastroenterol,2006,12(2):187-191.
[14]THU-HA N,BARBARA S,STANIM IRA K,et al.Characterization and molecular cloning of a heterodimeric beta-galactosidase from the probiotic strain Lactobacillus acidophilus R22[J].FEMS M icrobiol Lett,2007,269(1):136-144.
[15]NGUYEN T H,SPLECHTNA B,YAMABHAIM,etal.Cloning and expression of theβ-galactosidase genes from Lactobacillus reuteri in Escherichia coli[J].J Biotechnol,2007,129(4):581-591.
[16]侯重文,釗倩倩,劉飛,等.米曲霉乳糖酶在畢赤酵母中的高效表達[J].藥物生物技術,2014,21(1):22-25.
[17]符仙,陳麗萍,張愛聯,等.用畢赤酵母的GAP啟動子調控表達L-阿拉伯糖異構酶[J].工業微生物,2014,44(1):51-54.
Cloning of Lactobacillus kefiranofaciens β-galactosidase genes and expression in Pichia pastoris
XING Zhuqing,WANG Yanning,LIU Zhaoxian,WU Yanan,LIANG Li,WANG Yanping*
(College ofFood Engineeringand Biotechnology,Tianjin University ofScience&Technology,Tianjin 300457,China)
Using kum iss Lactobacillus kefiranofaciens ZW 3 genome as template,theβ-galactosidase genes(LacL and LacM)were am plified from L. kefiranofaciens by PCR,and itwas recombined into vector pPIC9K after digestion by EcoRIand SnaBI.Then two plasm ids of pPIC9K-LacL and pPIC9K-LacM were constructed through transformation into Escherichia coli DH5αand the nucleic acid sequencewasverified by sequencing results. The recombinationsof pPIC9K-LacL-GS115 and pPIC9K-LacM-GS115 were constructed by electroporating the vectors into Pichia pastoris GS115. Through screening,themulti-copy recombinations pPIC9K-LacL-GS115 and pPIC9K-LacM-GS115 were obtained resistant to 3.0 mg/m l G418. Through reverse PCR verification,LacL genes in recombination pPIC9K-LacL-GS115 were detected,in which fermented for 72 h induced by methanol.Moreover,theactivity ofβ-galactosidase from recombination pPIC9K-LacL-GS115 reached to 1.17 U/m l.A lso,the targetprotein band of recombination pPIC9K-LacM-GS115wasobserved by SDS-PAGE.
β-galactosidase;Pichia pastoris;clone;expression
Q556
0254-5071(2016)01-0010-04
10.11882/j.issn.0254-5071.2016.01.003
2015-11-16
國家自然科學基金資助項目(31171629);天津科技大學大學生實驗室創新基金項目(1414A303)
邢竹青(1989-),女,博士研究生,研究方向為食品科學。
王艷萍(1962-),女,教授,博士,研究方向為食品科學。
