加強表達3-磷酸甘油醛脫氫酶對谷氨酸棒桿菌產L-絲氨酸的影響
2016-10-13 00:43:15郭雯賴聯賀張曉梅史勁松許正宏
食品與發酵工業 2016年9期
郭雯,賴聯賀,張曉梅,史勁松,許正宏*
1(江南大學 藥學院,制藥工程研究室,江蘇 無錫,2141222)2(江南大學 生物工程學院,工業生物技術教育部重點實驗室,江蘇 無錫,214122)
?
加強表達3-磷酸甘油醛脫氫酶對谷氨酸棒桿菌產L-絲氨酸的影響
郭雯1,2,賴聯賀1,張曉梅1,史勁松1,許正宏1,2*
1(江南大學 藥學院,制藥工程研究室,江蘇 無錫,2141222)2(江南大學 生物工程學院,工業生物技術教育部重點實驗室,江蘇 無錫,214122)
在谷氨酸棒桿菌(Corynebacteriumglutamicum)中,由3-磷酸甘油醛脫氫酶(glyceraldehyde-3-phosphate dehydrogenase,GAPDH)催化的反應是糖酵解途徑的限速步驟,該反應還直接影響了L-絲氨酸的前體3-磷酸甘油酸的合成。研究首先比較了產L-絲氨酸的野生型菌株C.glutamicumSYPS-062與模式菌株C.glutamicumATCC14067的GAPDH酶活力,發現SYPS-062的GAPDH酶活力比ATCC14067高了55.8%。進一步采用在C.glutamicum33a△SS基因組上增加gapA拷貝數的方法加強表達GAPDH,構建了重組菌C.glutamicum33a△SS-2gapA。重組菌GAPDH轉錄水平和酶活力分別提高119%和53%,最大比生長速率提高10.6%,總糖耗速率提高4.4%,L-絲氨酸產量提高17.4%,糖酸轉化率提高12.2%,生產強度提高17.4%。結果表明,加強表達GAPDH能夠提高重組菌的生長和糖耗速率,并能夠提高L-絲氨酸的產量、糖酸轉化率和生產強度。
3-磷酸甘油醛脫氫酶;谷氨酸棒桿菌;L-絲氨酸
L-絲氨酸作為一種重要的氨基酸,參與很多重要代謝產物的合成,在醫藥、化妝品和食品添加劑領域具有廣泛的應用前景[1-2]。生產方法主要有化學合成法、蛋白質水解法、酶法轉化法和微生物發酵法。目前直接利用糖質原料的微生物發酵法受到了廣泛的關注。谷氨酸棒桿菌(Corynebacteriumglutamicum)作為一種食品安全級的菌株,擁有成熟的遺傳背景,已經廣泛應用于生產多種氨基酸[3]。
谷氨酸棒桿菌中L-絲氨酸的合成及降解途徑如圖1所示。L-絲氨酸以糖酵解途徑中間產物3-磷酸甘油酸作為起始物質,通過3-磷酸甘油酸脫氫酶(PGDH)、磷酸絲氨酸轉氨酶(PSAT)和磷酸絲氨酸磷酸酶(PSP)3個酶催化生成L-絲氨酸。L-絲氨酸則被絲氨酸羥甲基轉移酶(SHMT)或L-絲氨酸脫氨酶(L-serDH)催化降解為甘氨酸或丙酮酸[4]。目前代謝工程改造谷氨酸棒桿菌產L-絲氨酸主要集中在其合成及降解途徑。STOLZ等加強表達了C.glutamicumATCC13032L-絲氨酸合成途徑中的關鍵酶,敲除或弱化了降解途徑中的酶,菌株產量為345 mmol/L[5]。
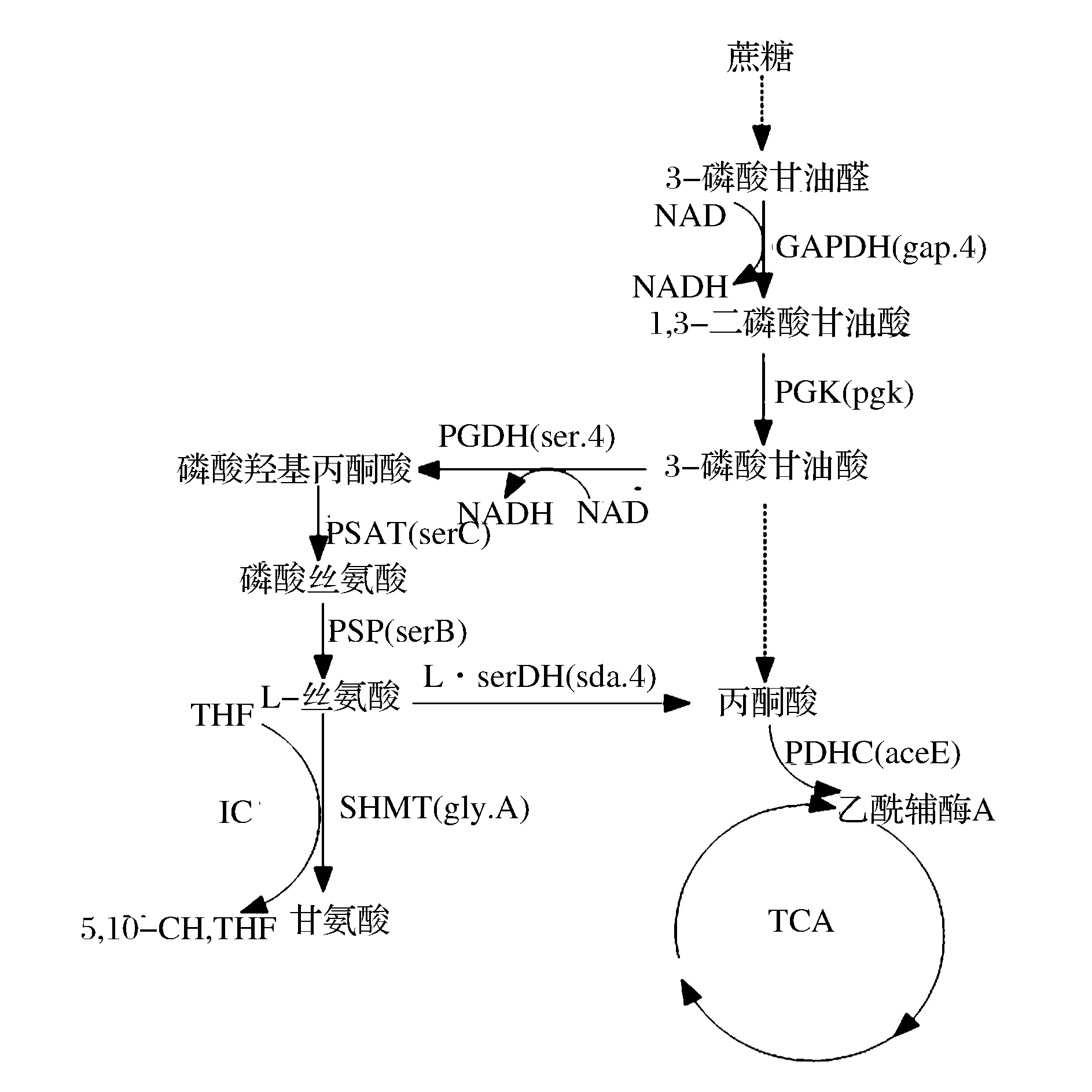
圖1 谷氨酸棒桿菌中L-絲氨酸的合成及降解途徑Fig.1 The biosynthesis and degradation pathway of L-serine in C. glutamicum
本實驗室前期對產L-絲氨酸的野生型菌株C.glutamicumSYPS-062進行了隨機誘變,得到了L-絲氨酸高產菌株C.glutamicumSYPS-062-33a。進而以其為出發菌株,對L-絲氨酸合成及降解途徑進行了改造,解除了L-絲氨酸對PGDH的反饋抑制,敲除了L-serDH,構建的重組菌C.glutamicum33a△SSL-絲氨酸產量為21.27 g/L[6]。目前針對糖酵解途徑的L-絲氨酸代謝工程改造策略則鮮有報道[7]。
3-磷酸甘油醛脫氫酶(glyceraldehyde-3-phosphate dehydrogenase,GAPDH)是糖酵解途徑中的關鍵酶,參與了L-絲氨酸前體3-磷酸甘油酸的合成。YAMAMOTO等通過加強表達GAPDH,提高了GAPDH酶活力,減少了胞內糖酵解途徑中3-磷酸甘油醛上游的中間代謝產物積累量,提高了谷氨酸棒桿菌在缺氧條件下的L-丙氨酸產量和糖耗速率[8]。GAPDH酶活力大小也可能會對谷氨酸棒桿菌產L-絲氨酸產生影響。
本文首先通過比較產L-絲氨酸的野生型菌株C.glutamicumSYPS-062與模式菌株C.glutamicumATCC14067的GAPDH酶活力,推測較高的GAPDH酶活力有利于L-絲氨酸的積累。進一步采用在C.glutamicum33a△SS基因組上增加gapA拷貝數的方法加強表達GAPDH,構建了重組菌C.glutamicum33a△SS-2gapA。最后通過比較出發菌和重組菌的發酵性能,評估了加強表達GAPDH對谷氨酸棒桿菌生長和產L-絲氨酸的影響。
1 材料與方法
1.1材料
1.1.1菌種及引物
C.glutamicumSYPS-062為本實驗室前期篩選并保藏的直接利用糖質原料產L-絲氨酸的野生型菌株;C.glutamicum33a△SS為本實驗室前期構建并保藏的產L-絲氨酸的重組菌;模式菌株C.glutamicumATCC14067、大腸桿菌JM109及穿梭質粒pk18mobsacB均為本實驗室保藏;本文所用的引物見表1。
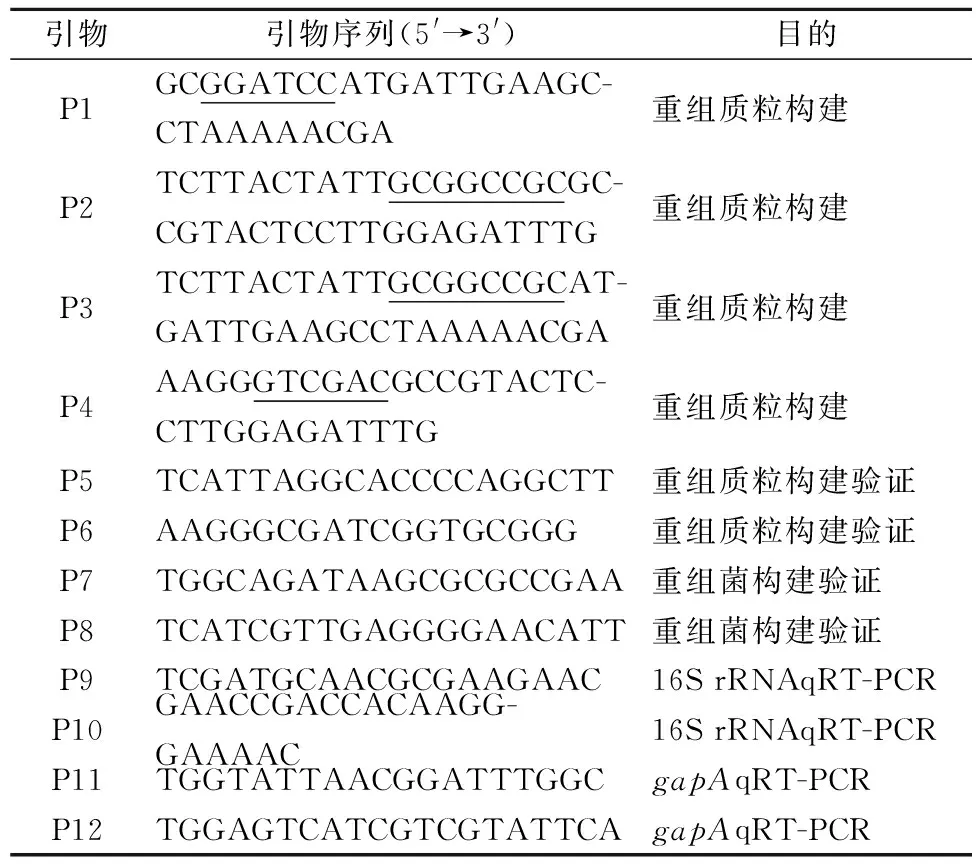
表1 本文所用的引物
注:酶切位點用下劃線標注。
1.1.2試劑
質粒小量提取試劑盒、膠回收試劑盒、柱式Trizol總RNA抽提試劑盒、抗生素、PCR引物等購自上海生工生物工程有限公司;PCR相關酶,限制性內切酶SalI、BamHI、NotI等購自TaKaRa公司;酶活測定試劑購自sigma公司;一步法cDNA合成試劑盒、Power qPCR PreMix(SYBR Green)購自上海捷瑞生物工程有限公司;其他試劑為國產分析純。
1.1.3培養基
(1) LB培養基(g/L):蛋白胨10;酵母粉 5;NaCl 10(固體培養基,瓊脂粉 20);滅菌條件:121 ℃,20 min。
(2)種子培養基(g/L):腦心浸液37;葡萄糖 20;(NH4)2SO410;MgSO4·7H2O 0.5;K2HPO40.2;NaH2PO40.3;滅菌條件:115 ℃, 7 min。
(3)發酵培養基(g/L):蔗糖 100;(NH4)2SO430;CaCO360;KH2PO43;MgSO4·7H2O 0.5;FeSO4·7H2O 0.02;MnSO4·H2O 0.02;原兒茶酸0.03;生物素 5×10-5;鹽酸硫胺素 4.5×10-4;滅菌條件:115 ℃,7 min。
(4)谷氨酸棒桿菌感受態培養基(g/L):蛋白胨 10;酵母粉 5;NaCl 10;吐溫80 1;甘氨酸 25;滅菌條件:121 ℃,20 min。
(6)谷氨酸棒桿菌電轉化培養基(g/L):蛋白胨 5;酵母粉 2.5;NaCl 5;腦心浸液 18.5;山梨醇 91;瓊脂粉 20;滅菌條件:121 ℃,20 min。
(7)10%蔗糖篩選培養基(g/L):腦心浸液37;蔗糖 100;(NH4)2SO410;MgSO4·7H2O 0.5;K2HPO40.2;NaH2PO40.3;瓊脂粉 20;滅菌條件:115 ℃,7 min。
1.2實驗方法
1.2.1重組質粒pk18mobsacB-2gapA的構建
在基因組上增加基因拷貝數的重組質粒構建方法參考文獻[9]。質粒構建流程圖如圖2所示。
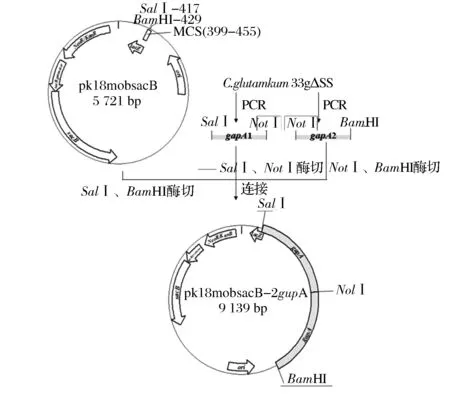
圖2 重組質粒pk18mobsacB-2gapA構建流程圖Fig.2 The flow chart of the construction of the recombinant plasmid pk18mobsacB-2gapA
以C.glutamicum33a△SS基因組為模板,采用引物P1/P2和P3/P4分別擴增含有不同酶切位點的片段gapA1和gapA2,片段總長度為1 709 bp,片段不僅包括gapA基因編碼區(1005 bp)還包括gapA基因與其上下游基因之間的間隔序列(上游間隔序列488 bp,下游間隔序列216 bp),使得擴增的片段涵蓋該基因的啟動子區和終止子區。將膠回收后的gapA1片段以BamHI和NotI雙酶切,gapA2片段以NotI和SalI雙酶切,并同時雙酶切質粒pk18mobsacB。膠回收酶切后的片段和質粒,T4DNA連接酶過夜連接。將連接產物化轉E.coliJM109,提取質粒,采用根據pK18mobsacB多克隆位點上下游序列設計的通用引物P5/P6進行PCR驗證,采用限制性內切酶BamHI和SalI進行酶切驗證。
1.2.2重組菌33a△SS-2gapA的構建
將成功構建的重組質粒pk18mobsacB-2gapA電轉化33a△SS感受態細胞[10],在含50 μg/mL Kan的平板中進行第1次同源重組[11]。挑取重組子在含Kan的種子培養基中過夜培養,稀釋涂布于10%蔗糖篩選培養基平板進行第2次同源重組。待長出單菌落后,挑取二次重組子,采用根據gapA上下游基因的編碼區設計的引物P7/P8進行菌落PCR驗證。
1.2.3GAPDH酶活力的測定
取培養至對數生長期的種子或發酵液10 mL離心,用100 mmol/L Tris-HCl洗滌菌體3次。用1 mL的Tris-HCl重懸菌體,在冰浴條件下超聲破碎45 min (破3 s,停7 s)。離心去除細胞碎片得到上清液即為粗酶液,再采用Bradford蛋白定量試劑盒[12]進行蛋白濃度定量。
酶活力測定方法參考文獻[8]。GAPDH酶活在25 mmol/L磷酸鹽緩沖液(pH 7.5)中進行測定,體系中包括0.2 mmol/L EDTA,5 mmol/L NAD+,5 mmol/L 3-磷酸甘油醛。
1單位酶活力定義:在37 ℃、pH 7.5的反應條件下,每分鐘生成1 μmol NADH所需的酶量,(NADH μmol/min)。其中NADH摩爾吸光系數采用6.22×103M-1·cm-1[8]。
比活力=酶液稀釋倍數×ΔA340×Vt/(e×Vs×d×C)
式中:ΔA340為吸光度值變化率,min-1;Vt為反應體系總體積,mL;e為摩爾吸光系數,M-1·cm-1;Vs為粗酶液體積,mL;d為比色杯光徑,cm;C為蛋白的質量濃度,mg/mL。
1.2.4GAPDH相對轉錄水平的測定
總RNA提取按柱式Trizol總RNA抽提試劑盒說明書進行。將重組菌和出發菌的RNA定量成相同濃度后進行逆轉錄。以重組菌和出發菌的cDNA為模板進行RT-qPCR。16S rRNART-qPCR采用引物P9/P10,gapART-qPCR采用引物P11/P12。以16S rRNA的Ct值作為內參計算GAPDH的轉錄水平。RT-qPCR反應體系如下:2×Power qPCR PreMix 10 μL,ROX Reference Dye(50×) 0.4 μL,cDNA模板2 μL,引物(Forward +Reverse) 4 μL,ddH2O 3.6 μL,總體積20 μL。反應程序如下:95 ℃ 30 s;95 ℃ 5 s,60 ℃ 30 s,72 ℃ 45 s,循環40次。每個樣品重復3次。RT-qPCR結果采用2-△△Ct方法[13]對基因的Ct值進行定量分析。
1.2.5發酵參數的測定
生物量的測定:紫外分光光度計測定OD562值;DCW (g/L) = 0.192 5×OD562。
糖和氨基酸濃度的測定:采用高效液相色譜法(HPLC)測定[14]。
2 結果與分析
2.1產L-絲氨酸的野生型菌株與模式菌株GAPDH酶活力的比較
取對數生長期的ATCC14067與SYPS-062種子液,測定GAPDH酶活力,測定結果如圖3 (A)所示,SYPS-062的GAPDH酶活力為(0.804±0.006) U/mg,比ATCC14067[(0.516±0.005) U/mg]高了55.8%。取同一批種子液,比較SYPS-062與ATCC14067的GAPDH轉錄水平,結果如圖3 (B)所示,SYPS-062的GAPDH轉錄水平為ATCC14067的91.9%,在轉錄水平上未見明顯差異。
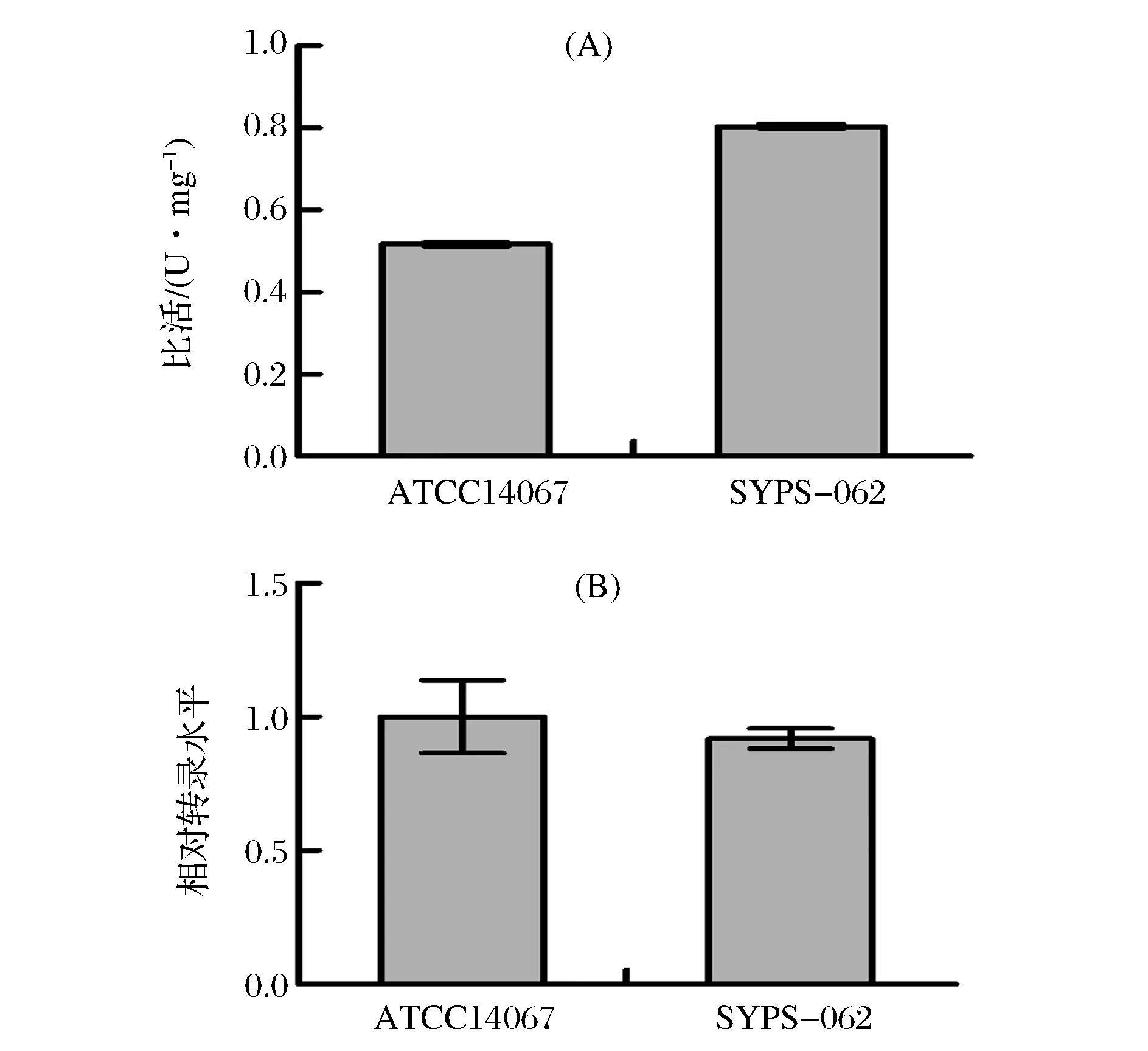
圖3 ATCC14067與SYPS-062中GAPDH酶活力和轉錄水平比較Fig.3 The specific enzyme activity and relative transcriptional level comparisons of GAPDH between ATCC14067 and SYPS-062
進一步比較C.glutamicumSYPS-062(GenBank accession: JXBH00000000)與C.glutamicumATCC14067(GenBank accession: AGQQ00000000)GAPDH的核糖體結合位點(RBS)[15]和編碼區序列,發現兩者GAPDH的RBS序列一致;在GAPDH編碼區存在1個氨基酸序列的差異,第249位氨基酸在ATCCC14067中為蘇氨酸,而在SYPS-062中為異亮氨酸。于是推測ATCC14067與SYPS-062的GAPDH酶活力差異可能是由它們GAPDH編碼區的氨基酸序列差異所致。鑒于ATCC14067無法積累L-絲氨酸,而SYPS-062能產L-絲氨酸,推測較高的GAPDH酶活力有利于L-絲氨酸積累,擬在菌株C.glutamicum33a△SS上進一步加強表達GAPDH,以期提高L-絲氨酸的產量。
2.2重組菌與出發菌GAPDH相對轉錄水平的測定
采用1.2.1與1.2.2中的方法,成功構建了重組質粒pk18mobsacB-2gapA與重組菌C.glutamicum33a△SS-2gapA。取對數生長期的重組菌和出發菌發酵液,測定GAPDH相對轉錄水平,測定結果如圖4所示。從圖4可以看出重組菌GAPDH轉錄水平為出發菌的2.19倍。在基因組上增加gapA拷貝數后重組菌GAPDH的轉錄水平得到了提高。
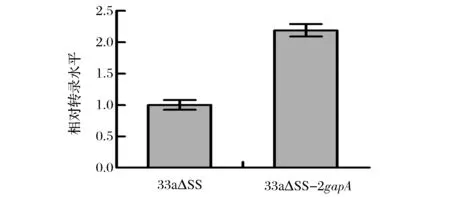
圖4 33a△SS和33a△SS-2gapAGAPDH相對轉錄水平的測定Fig.4 Therelative transcriptional level of GAPDH in 33a△SS and 33a△SS-2gapA
2.3重組菌與出發菌GAPDH酶活力的測定
取對數生長期的重組菌和出發菌發酵液,測定GAPDH酶活力,測定結果如圖5所示。重組菌GAPDH酶活力為(1.052±0.025) U/mg,為出發菌[(0.688±0.005) U/mg]的1.53倍。在基因組上增加gapA拷貝數后重組菌GAPDH的酶活力提高。
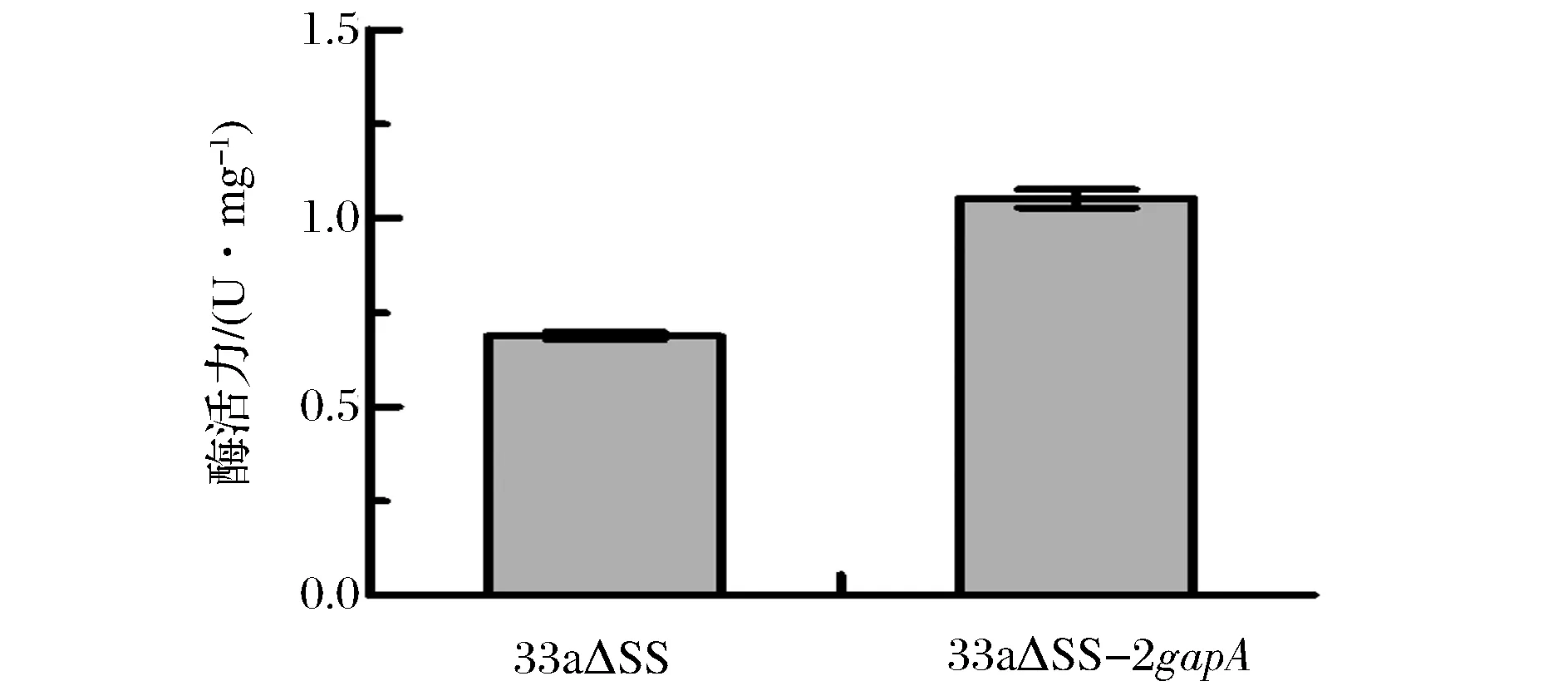
圖5 33a△SS和33a△SS-2gapA GAPDH酶活力測定Fig.5 The specific activity of GAPDH in 33a△SS and 33a△SS-2gapA
2.4重組菌發酵性能評價
在以100 g/L蔗糖為底物的發酵培養基中比較重組菌和對照菌的發酵性能。菌體的生長曲線如圖6 (A)所示,從圖6 (A)可以看出,重組菌的生長速率在24 h~60 h期間明顯快于出發菌,發酵至84 h重組菌OD達到最大值51.40,其最大比生長速率為0.209 h-1,比出發菌(0.189 h-1)提高了10.6%,可見加強表達GAPDH能夠提高重組菌的生長速率。菌體的糖耗曲線如圖6 (B)所示,可以看出,在菌體的對數生長期(24 h~48 h)重組菌的糖耗速率明顯快于出發菌,發酵至84h重組菌糖消耗完畢,其總糖耗速率為1.18 g/(L·h),比出發菌(1.13 g/(L·h))提高了4.4%,可見加強表達GAPDH能夠提高重組菌的糖耗速率。菌體的產酸曲線如圖6 (C)所示,從圖6 (C)可以看出,發酵至84h重組菌的L-絲氨酸產量達到最大值23.73 g/L,比出發菌(20.21 g/L)提高了17.4%;重組菌糖酸轉化率達0.239 g/g 蔗糖,比出發菌(0.213 g/g 蔗糖)提高了12.2%;重組菌生產強度達0.283 g/(L·h),比出發菌[0.241 g/(L·h)]提高了17.4%;可見加強表達GAPDH能夠提高重組菌的產量、糖酸轉化率和生產強度。重組菌和出發菌84 h時的發酵參數如表2所示。綜上所述,加強表達GAPDH的代謝工程策略能夠獲取更高效合成L-絲氨酸的重組菌株。
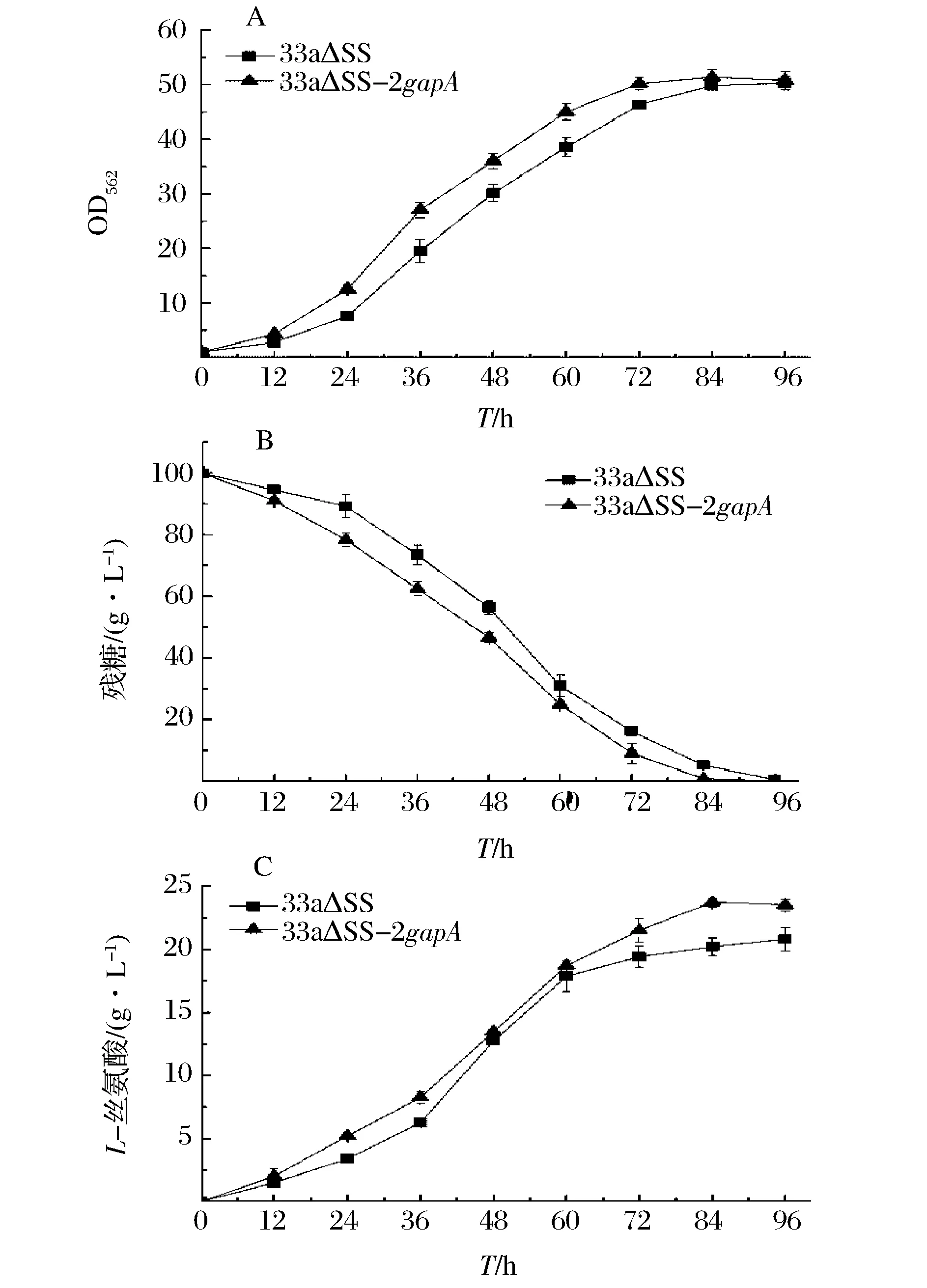
(A) 生長曲線; (B) 糖消耗曲線; (C) L-絲氨酸生產曲線圖6 出發菌33a△SS和重組菌33a△SS-2gapA發酵性能比較Fig.6 The fermentation performancecomparison of 33a△SS and 33a△SS-2gapA
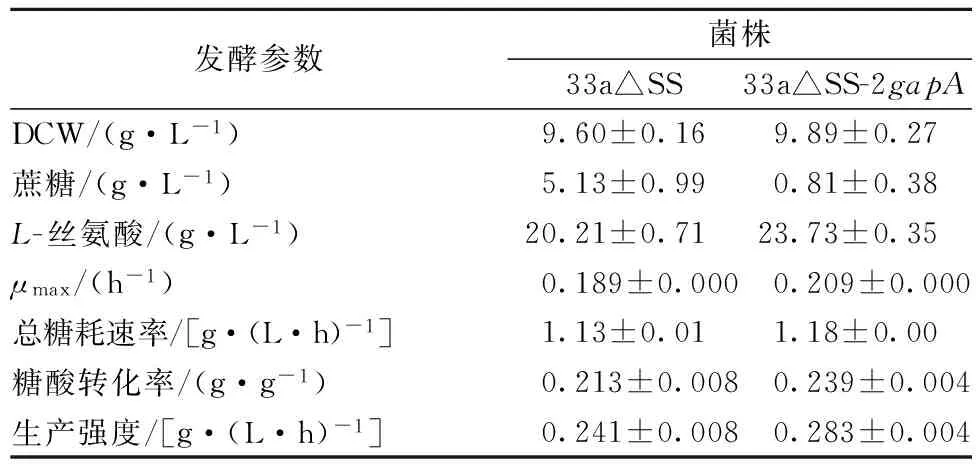
發酵參數菌株33a△SS33a△SS-2gapADCW/(g·L-1)9.60±0.169.89±0.27蔗糖/(g·L-1)5.13±0.990.81±0.38L-絲氨酸/(g·L-1)20.21±0.7123.73±0.35μmax/(h-1)0.189±0.0000.209±0.000總糖耗速率/[g·(L·h)-1]1.13±0.011.18±0.00糖酸轉化率/(g·g-1)0.213±0.0080.239±0.004生產強度/[g·(L·h)-1]0.241±0.0080.283±0.004
3 結論
本文首先比較了產L-絲氨酸的野生型菌株C.glutamicumSYPS-062與模式菌株C.glutamicumATCC14067的GAPDH酶活力,發現SYPS-062的GAPDH酶活力比ATCC14067高了55.8%。兩者的GAPDH編碼區存在一個氨基酸序列差異,推測酶活差異是由該氨基酸序列差異所致,進一步證明還有待后續實驗對該酶進行酶學性質研究。鑒于ATCC14067無法積累L-絲氨酸,而SYPS-062能產L-絲氨酸,推測較高的GAPDH酶活力有利于L-絲氨酸積累。進而以C.glutamicum33a△SS為出發菌株,在其基因組上增加了1個gapA的拷貝數,構建了加強表達GAPDH的重組菌C.glutamicum33a△SS-2gapA,并考察了該酶的加強表達對谷氨酸棒桿菌產L-絲氨酸的影響。鑒于質粒加強表達的不穩定性及需要在發酵過程中添加抗生素等缺點,采用在基因組上增加gapA拷貝數的方法加強表達GAPDH。比較重組菌和出發菌中GAPDH的轉錄水平和酶活力,重組菌GAPDH轉錄水平和酶活力分別提高119%和53%。發酵實驗結果表明,重組菌最大比生長速率提高10.6%,糖耗速率提高4.4%,L-絲氨酸產量提高17.4%,糖酸轉化率提高12.2%,生產強度提高17.4%。可見加強表達GAPDH能夠提高重組菌的生長和糖耗速率,并能夠提高L-絲氨酸的產量、糖酸轉化率和生產強度。本文首次報道了加強表達GAPDH的代謝工程策略能夠獲取更高效合成L-絲氨酸的重組菌株。
[1]GU Peng-fei, YANG Fan, SU Tian-yuan, et al. Construction of anL-serine producingEscherichiacolivia metabolic engineering [J]. Journal of Industrial Microbiology & Biotechnology,2014, 41(9):1 443-1 450.
[2]ZHANG Xiao-mei, XU Guo-qiang, LI Hui, et al. Effect of cofactor folate on the growth ofCorynebacteriumglutamicumSYPS-062 andL-serine accumulation [J]. Applied Biochemistry and Biotechnology,2014, 173(7):1 607-1 617.
[3]VERTES A A, INUI M, YUKAWA H. Postgenomic approaches to using corynebacteria as biocatalysts [J]. Annual Review of Microbiology,2012, 66:521-550.
[4]PETERS-WENDISCH P, NETZER R, EGGELING L, et al. 3-Phosphoglycerate dehydrogenase fromCorynebacteriumglutamicum: the C-terminal domain is not essential for activity but is required for inhibition byL-serine [J]. Applied Microbiology and Biotechnology, 2002, 60(4):437-441.
[5]STOLZ M, PETERS-WENDISCH P, ETTERICH H, et al. Reduced folate supply as a key to enhancedL-serine production byCorynebacteriumglutamicum[J]. Applied Environmental Microbiology,2007, 73(3):750-755.
[6]XU Guo-qiang, ZHU Qin-jian, LUO Yu-chang, ZHANG Xiao-mei, et al. Enhanced production ofL-serine by deletingsdaAcombined with modifying and overexpressingserAin a mutant ofCorynebacteriumglutamicumSYPS-062 from sucrose [J]. Biochemical Engineering Journal,2015, 103:60-67.
[7]LAI Shu-juan, ZHANG Yun, LIU Shu-wen, et al. Metabolic engineering and flux analysis ofCorynebacteriumglutamicumforL-serine production [J]. Sci China Life Sci,2012, 55(4):283-290.
[8]YAMAMOTO S, GUNJI W, SUZUKI H, et al. Overexpression of genes encoding glycolytic enzymes inCorynebacteriumglutamicumenhances glucose metabolism and alanine production under oxygen deprivation conditions [J]. Applied Environmental Microbiology, 2012, 78(12):4 447-4 457.
[9]BECHER J, ZELDER O, HAFNER S, et al. From zero to herodesign-based systems metabolic engineering ofCorynebacteriumglutamicumforL-lysine production [J]. Metabolic Engineering,2011, 13(2):159-168.
[10]VAN DER REST ME, LANGE C, et al.A heat shock following electroporation induces highly efficient transformation ofCorynebacteriumglutamicumwith xenogeneic plasmid DNA [J]. Applied Microbiology Biotechnology, 1999, 52(4):541-545.
[11]SCHAFER A, TAUCH A, JAGER W, et al. Small mobilizable multi-purpose cloning vectors derived from theEscherichiacoliplasmids pK18 and pK19: selection of defined deletions in the chromosome ofCorynebacteriumglutamicum[J]. Gene, 1994, 145(1):69-73.
[12]BRADFORD MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding [J]. Analytical Biochemistry, 1976, 72:248-254.
[13]LIVAK KJ, SCHMITTGEN TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method [J]. Methods, 2001, 25(4):402-408.
[14]ZHU Qin-jian, ZHANG Xiao-mei, LUO Yu-chang, et al.L-Serine overproduction with minimization of by-product synthesis by engineeredCorynebacteriumglutamicum[J]. Applied Microbiology Biotechnology,2015, 99(4):1 665-1 673.
[15]KOICHI T, HARUHIKO T, MASAYUKI I, et al. Involvement of the LuxR-Type transcriptional regulatorramA in regulation of expression of thegapA gene, encoding glyceraldehyde-3-phosphate dehydrogenase ofCorynebacteriumglutamicum[J]. Journal of Bacteriology, 2009, 191(3): 968-977.
Effects of glyceraldehyde-3-phosphate dehydrogenase overexpression onL-serine production inCorynebacteriumglutamicum
GUO Wen1,2, LAI Lian-he1, ZHANG Xiao-mei1, SHI Jin-song1, XU Zheng-hong1,2*
1(Laboratory of Pharmaceutical Engineering, School of Pharmaceutics Science, Jiangnan University, Wuxi 214122, China)2 (The Key Laboratory of Industrial Biotechnology, Ministry of Education, School of Biotechnology, Jiangnan University, Wuxi 214122, China)
InCorynebacteriumglutamicum, the reaction catalyzed by glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is the rate-limiting step in the glycolysis pathway, also, it influences the biosynthesis of theL-serine precursor 3-phosphoglycerare directly. In this study, firstly, the GAPDH activity of the wild-typeL-serine-producing strainC.glutamicumSYPS-062 and the model strainC.glutamicumATCC14067 were compared. The GAPDH activity in SYPS-062 was 55.8% higher than that in ATCC14067. Then, GAPDH was overexpressed by insertion of an additional gapA copy into genome ofC.glutamicum33a△SS to construct the recombinant strainC.glutamicum33a△SS-2gapA. The transcriptional level and the enzyme activity of GAPDH in the recombinant strain were increased by 119% and 53% respectively. The fermentation experiment showed that the maximum specific growth rate was increased by 10.6%, the sugar consumption rate was increased by 4.4%, theL-serine production was increased by 17.4%, the yield was increased by 12.2%, and the productivity was increased by 17.4% in the recombinant strain. These results demonstrated that overexpression of GAPDH could increase the growth and sugar consumption rate, and improve the production, yield and productivity ofL-serine in the recombinant strain.
glyceraldehyde-3-phosphate dehydrogenase;Corynebacteriumglutamicum;L-serine
10.13995/j.cnki.11-1802/ts.201609003
碩士研究生(許正宏教授為通訊作者,E-mail:zhenghxu@jiangnan.edu.cn)。
2016-03-08,改回日期:2016-03-18
