多重標準曲線技術在原子吸收光譜法測定陶瓷鉛、鎘溶出量中的應用
2016-12-13 09:27:01李文杰董一軍王海濤
中國陶瓷工業 2016年5期
關鍵詞:標準
李文杰,董一軍,王海濤
(唐山出入境檢驗檢疫局國家陶瓷檢測重點實驗室,河北 唐山 063000)
多重標準曲線技術在原子吸收光譜法測定陶瓷鉛、鎘溶出量中的應用
李文杰,董一軍,王海濤
(唐山出入境檢驗檢疫局國家陶瓷檢測重點實驗室,河北 唐山 063000)
簡述了火焰原子吸收光譜法測定日用陶瓷鉛、鎘溶出量及多重標準曲線法的基本原理,根據12組標準樣液不同的鉛、鎘濃度范圍,從一系列線性擬合回歸方程中尋找|A0-?0|最小者作為多重標準曲線法測定條件。比較其與國標法及常規法三種不同標準工作曲線條件下,同一樣液中重金屬濃度測定結果的差異程度并分析原因。推導出當鉛、鎘濃度低于40.0 mg/L和4.00 mg/L時,其測定結果準確性較高,但對中、高濃度樣液的測試準確度偏低。常規方法對濃度介于0.0 mg/L-1.0 mg/L和0.00 mg/L-0.10 mg/L之間樣液的測試誤差較大,對中、高濃度樣液的測試誤差較小。而多重標準曲線法對所有濃度樣液的鉛、鎘濃度檢測結果不確定度均最小。
多重標準曲線法;原子吸收光譜法;測定;日用陶瓷;鉛、鎘溶出量
0 引 言
近年來,鉛、鎘等重金屬中毒導致的食品安全事件頻頻發生,迫使人們將目光聚焦耕地、工業等重金屬污染高發領域。殊不知,作為與食品直接接觸的碗盤杯碟、餐茶酒具等陶瓷制品同樣存在著不容忽視的鉛、鎘超標隱患[1]。因在日用陶瓷生產制造過程中,基于助熔、著色等目的,釉料及顏料中可能含有一定量的鉛、鎘等重金屬元素[2]。在特定的接觸條件下,產品若用于盛裝酸性食物,則極易導致鉛、鎘溶出并遷移至食物從而最終進入人體[3]。而重金屬元素在人體內經過長期富集勢必對生命健康構成嚴重威脅,其中鉛對人體各類器官、組織均具有一定的蓄積性和親和性,可毒害神經、消化、造血等系統[4];進入人體的鎘主要累積于肝、腎、胰腺和骨骼中,會造成貧血、神經痛、骨質松軟和分泌失調等病癥,且治療極為困難[5]。鑒于鉛、鎘對人體危害的不可逆轉性,世界各國政府通常將盛裝和儲存食品的陶瓷器皿安全衛生問題等同于食品安全,對其鉛、鎘溶出量嚴加限制,并將之作為檢驗判定陶瓷制品合格與否的決定性指標。
目前,“日用陶瓷藝術化、藝術陶瓷日用化”的產品設計趨勢所帶來的高鉛、鎘含量花紙源頭控制監測需求和美國《加州65準則》[6]對日用陶瓷重金屬溶出量的超低限量規定,迫使檢測機構不得不同時處理各個濃度陶瓷鉛、鎘溶出量的檢測業務。實驗人員通常按照國標GB/T 3534-2002[7]和美標ASTM C 738-94(2011)[8]規定選擇特定濃度系列的標準溶液建立相應的標準曲線,然后在相同的工作曲線條件下連續進行大量不同濃度的陶瓷樣品鉛、鎘溶出量檢測;當樣品濃度跨度較大時,僅依靠原子吸收分光光度計在一定濃度范圍內采用二次方程進行曲線擬合進行分析計算必然造成較大的測量誤差。另外,火焰原子吸收光譜法的分析特點也決定了在不同濃度鉛、鎘元素含量測量過程中,按照上述標準規范要求所繪標準工作曲線的有效測量范圍相對狹窄,一般以0.2-0.8 Abs為限。同時,采用常規方法使標準曲線覆蓋待測樣品整個濃度范圍時,由于高濃度溶液在原子化器中生成的基態原子不成比例,將使校準曲線產生彎曲;且伴隨著溶液中待測組分濃度的增加,粒子的摩爾吸光系數也將最終發生變化,從而導致較大的測試誤差。因此,為提高陶瓷樣品重金屬溶出量測定結果的準確性,本文通過理論分析和實驗驗證的方式研究采取多重標準曲線技術運用最小二乘法將整個樣品濃度范圍內的標準系列分段進行曲線擬合,探索與高、中、低不同重金屬溶出水平相對應的最佳標準曲線的選擇途徑,力求實現測量誤差最小化。
1 測試原理
1.1 原子吸收光譜法測定日用陶瓷鉛、鎘溶出量的基本原理
原子吸收光譜法是在待測元素的特定波長下,通過測量試樣所產生的原子蒸氣對輻射的吸收來測定其該元素濃度的方法。當元素原子被外界能量激發時,處于基態的電子吸收一定能量后以某種概率躍遷到較高能級的激發態;當處于不穩態的高能電子通過發射某種波長光的形式釋放能量恢復穩定時便產生了原子光譜。電子在基態和最鄰近于基態的激發態之間的躍遷所產生的譜線稱為共振線,原子吸收光譜分析基于共振躍遷的可逆性原理和不同元素共振線的唯一性特征,通過測量處于基態的原子蒸氣對某一特征譜線的吸收量值分析確定其原子含量[9]。
根據Lanbert_Beer定律,當強度為I0、頻率為ν的光源透過厚度為l的原子蒸氣后,經共振吸收[10],透過的光強Iν可表示為Iν=I0eKνl,對其進行變形得到lg(Iν/ I0)=-0.434 Kνl;令lg(Iν/ I0)= A,對于類似空心陰極燈的銳線光源可用K0代替Kν,故A=lg(Iν/ I0)=0.434 K0l=KNl;式中,A為吸光度,K為常數,N為原子總數,l為原子蒸氣的厚度(儀器火焰寬度)。由此可見,吸光度分別與原子總數(溶液中原子的濃度)和原子蒸氣的厚度成正比例關系。
1.2 多重標準曲線技術原理
標準曲線是標準物質的物理/化學屬性跟儀器響應之間的函數關系,標準曲線法是原子吸收光譜分析中最常用方法之一[11]。通過配制與待測試樣基體相同的含有不同濃度待測組分的一系列標準溶液,以空白溶液為參比溶液,在選定的波長下,分別測定其吸光度。然后以標準溶液濃度為橫坐標,吸光度為縱坐標繪制標準工作曲線。試樣經適當處理后,在完全相同的實驗條件下測量其吸光度。據此在標準曲線上查出樣液中被測元素的含量。
假設濃度為C1、C2、…Cn的N個標準溶液,其對應的吸光度測量值分別為A1、A2、...An,通常可采用最小二乘法進行多項式擬合,即

則上述多項式回歸問題可轉化為多元線性回歸問題來解決。由最小二乘法可得:
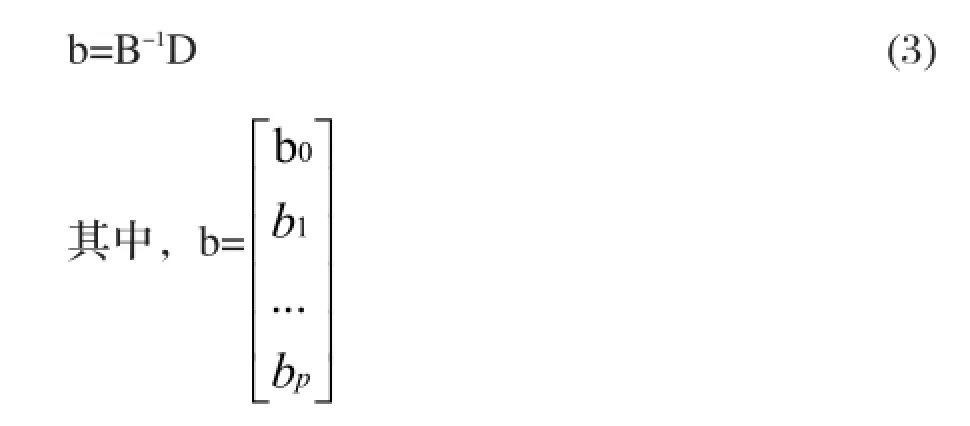

實驗的總偏差平方和為
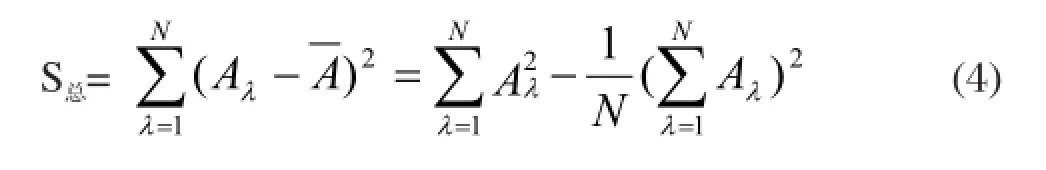
回歸方程的回歸平方和為
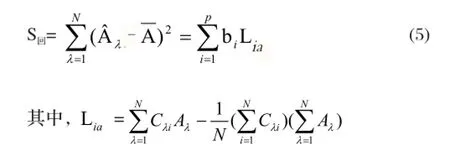
剩余平方和為
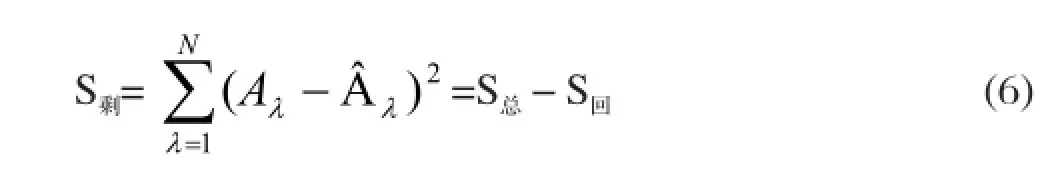
由統計量F回可檢驗回歸方程有無顯著意義,其值為

若F回>Fα(P,N-P-1),則認為線性回歸方程是有顯著意義的;其中,Fα(P,N-P-1)是在給定的顯著性水平α下的臨界值。由某一樣品的C01,C02,…,C0p值,可用回歸方程求得回歸值?0,它是該樣品測量值A0的估計值,偏差A0-?0服從正態分布,而且
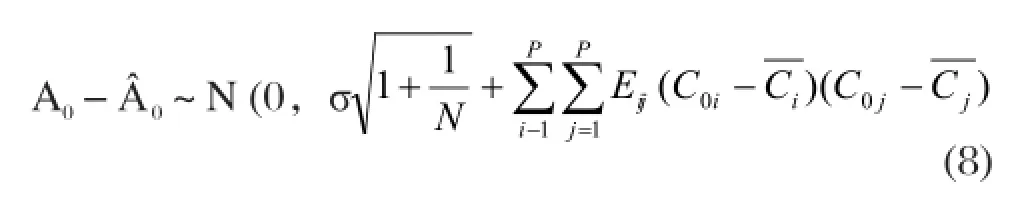
式中,Eij為相關矩陣E=B-1的元素,σ可由剩余平方和估計,即

由式(8)知,C01,C02,...,C0p偏離的值愈大,則值愈大。即在附近,利用回歸方程計算得到的結果精度最佳,而在偏離較大的低濃度或高濃度區間,即使測量誤差相同,由回歸方程計算得出的結果精度也較差。顯然,采用常規標準曲線法,在整個濃度范圍內進行擬合,存在較大的弊病;若采用多重標準曲線技術,將N個標準所測的結果分段進行擬合,得到一系列濃度范圍較窄的標準曲線。然后,根據樣液中待測元素含量,選擇最適宜的標準曲線計算樣液的精確濃度,可提高分析計算結果的精密度和準確度。
2 實驗部分
2.1 儀器與試劑
AA240FS原子吸收分光光度計(美國Varian公司、配套鉛、鎘空心陰極燈);XMD型智能溫度巡檢儀(上海普躍電氣有限公司);陶瓷浸泡柜(宜興市揚子江環保實驗設備廠,專利號:200920062955);鉛、鎘國家標準溶液(中國計量科學研究院, 1000 μg/ml);冰乙酸(分析純, 密度為1.05 g/cm3, 天津市富宇精細化工有限公司);實驗用水由Synery UV超純水系統(美國Millipore公司)制備。
2.2 儀器工作條件
陶瓷樣品中鉛、鎘溶出量采用火焰原子吸收分光光度計進行測定,儀器工作條件見表1。
2.3 樣品采集及前處理
為確保實驗樣品的代表性和均勻性,特向相關能力驗證提供者購買數件濃度范圍為0.01mg/ L~10.00mg/L的日用陶瓷釉面鉛、鎘溶出量檢測標準樣品和標準樣液,然后依據標準ASTM C 738-94(2011)要求對其進行濃縮處理。即準確移取50.0 ml萃取液于250 ml的燒杯中,在蒸汽浴中蒸發至近干,然后加入1 mlHCL蒸發至干狀。精確添加樣液移取體積0.1倍的4%醋酸,直至完全溶解余渣,使最終濃縮樣液中重金屬含量水平滿足實驗要求。并對所有樣品萃取溶液和標準樣液濃縮液進行有效標識。
對待測陶瓷樣品進行前處理時,首先將樣品在弱堿性洗滌液中清洗干凈,然后在自來水下反復沖洗并用超純水漂洗干凈, 于通風處晾干。
2.4 樣品浸泡及萃取液提取
在22±2 ℃的環境溫度條件下,將4%乙酸溶液順序注入54件樣品直至溢出口沿,浸泡24h±20 min后,使用硼硅質玻璃棒攪拌樣液使之混和均勻,再將所有萃取液移入樣品試劑瓶中保存并標記。
2.5 標準溶液的配制及鉛、鎘溶出量測定與計算
依據國標GB/T 3534-2002規定,分別配制濃度為0.0、0.5、1.0、2.0、3.0、4.0、5.0、6.0、7.0 mg/L和0.00、0.05、0.10、0.20、0.30、0.40、0.50、0.60、0.70 mg/L的鉛、鎘系列標準溶液,并據此繪制相應的標準曲線。其中,鉛元素的特征濃度為0.19 mg/L,曲線方程為A =0.32191×C2-0.07183×C+0.02408,線性相關系數r=1.0000;鎘元素的特征濃度為0.011 mg/L,曲線方程為A =1.41890×C2-0.62701×C+0.40910,線性相關系數r=1.0000。

表1 儀器工作條件(FAAS)Tab.1 Test conditions of FAAS
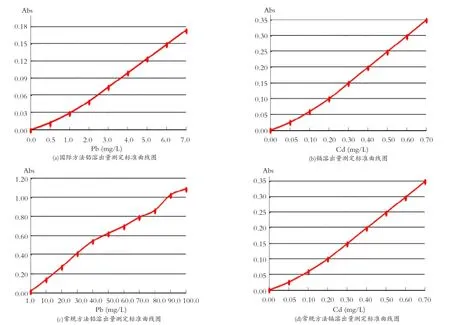
圖1 國標法和常規法鉛、鎘溶出量測定標準曲線示意圖Fig.1 The standard curve schematic diagram of lead and cadmium dissolution test by national standard method and conventional method
為客觀體現在待測樣品整體濃度范圍內利用二次方程進行曲線擬合的有效性,按照當前陶瓷產品釉面鉛、鎘溶出量的實際水平及花紙試燒等源頭控制的檢測需求,分別以1.0、10.0、20.0、30.0、40.0、50.0、60.0、70.0、80.0、90.0、100.0mg/L和0.10、1.00、2.00、3.00、4.00、5.00、6.00、7.00、8.00、9.00、10.00 mg/L作為鉛、鎘系列標準溶液,并據此繪制相應的常規標準曲線。其中,鉛元素的特征濃度為7.63mg/L,曲線方程為A= -0.40549×C2+0.01604×C+0.00098,線性相關系數r=0.9994;鎘元素的特征濃度為0.047mg/ L,曲線方程為A =5.82976×C2-0.79069×C+ 0.17628,線性相關系數r=0.9999。上述國標及常規標準曲線走勢圖如圖1所示。

表2 12組樣液所對應的最佳標準曲線方程及線性相關系數Tab.2 The contrast table of the best standard curve equation and linearly dependent coefficient in correspondence with 12 groups of sample liquid
為確保實現不同濃度區間樣液測定不確定度的最小化,須以儀器響應的特異性和標液與樣品的匹配性為前提繪制標曲。在擴展所需其他相關濃度系列標準溶液配制的基礎上,首先根據既定實驗樣液濃度范圍確定中間點,然后按照奇數點的設置原則,以此為中心左右對稱地分布標準曲線點。基于朗伯—比爾(Lanbert-Beer)定律和多重標準曲線技術原理,根據上述12組待測樣液鉛、鎘含量的既定濃度范圍,選取與其最接近的標準溶液濃度點。然后從該濃度附近的一系列線性擬合回歸方程中尋找能夠使|A0-?0|最小者[12],并以之為基礎繪制標準曲線。其線性擬合方程式及相關系數如表2所示。

表3 不同標準曲線條件下各濃度范圍內樣液測定值及誤差對比表Tab.3 The contrast table of testing results and measurement error of the concentration range of sample liquid under different standard curve conditions
3 結果與討論
為研究高、中、低不同濃度范圍的日用陶瓷鉛、鎘萃取樣液運用原子吸收光譜法進行測定時,采用不同濃度系列標準溶液所繪標準工作曲線對其檢測結果準確性的影響。基于國標所設標準曲線適用范圍的有限性,在按照美標ASTM C 738-94(2011)要求對鉛、鎘濃度超過7.0 mg/L和0.70 mg/L的樣液采取特定稀釋處理措施后,首先利用國標規定的標準曲線對12組原始樣液及其稀釋溶液進行鉛、鎘濃度測試,然后以常規標準曲線為基礎對上述原始樣液統一實施鉛、鎘濃度測定。同時依據各組樣液濃度范圍,采用表2所示多重標準工作曲線對其進行鉛、鎘濃度測定與計算。并將各項檢測結果與其已知真實濃度進行誤差分析,相關統計數據及不同標準曲線條件下各組樣液鉛、鎘濃度測定結果對比圖詳見表3和圖2。
由表3數據和圖2曲線走勢圖可知,鉛、鎘濃度范圍為0.5 mg/L-96.86 mg/L和0.060 mg/L-9.630 mg/L的12組樣液分別采用國標方法、常規方法和多重標準曲線法測定后,發現同一樣液在不同標準曲線測定條件下,其測試誤差大小及分布情況各異。當鉛、鎘濃度分別低于40.0 mg/L和4.00 mg/L時國標方法測定結果的準確性較高。但中、高濃度樣液在稀釋、蒸發和原子化過程中由于密度、黏度、表面張力等物理性質的變化將影響霧滴的細度、脫溶劑效率和蒸發效率,最終引起原子吸收信號強度的變化并直接或間接地導致原子化效率的改變。同時,樣液中存在的大量基體元素在火焰中蒸發解離時不僅需消耗大量的熱量,而且蒸發過程中有可能包裹待測元素,從而延緩待測元素的蒸發并降低原子化效率,使得鉛、鎘濃度測試誤差最高達到9.62%和17.57%。
而常規方法由于在較寬的濃度跨度和有限的標準點設置情況下,盡管濃度點的分布相對均勻,但標準曲線未能有效覆蓋鉛、鎘濃度區間介于0.0 mg/L-1.0 mg/L和0.00 mg/L-0.10 mg/L內的樣液。導致低濃度樣液測試結果誤差較大,因較低濃度與中、高濃度所提供信息量的微弱差異導致其鉛、鎘濃度測試誤差呈現36.54%、33.87%與0.44%、0.98%的較大偏離。鑒于多重標準曲線法根據待測樣液的大致濃度范圍,有針對性地選擇工作曲線重心點,并以之為中心左右對稱地分布濃度點,使得在標準曲線覆蓋的整個濃度范圍內對所有濃度的樣本提供的信息量等同。因此,鉛、鎘濃度檢測結果的不確定度最小,分別為5.77%和2.44%且測試結果與真實值二者之間曲線高度重合。
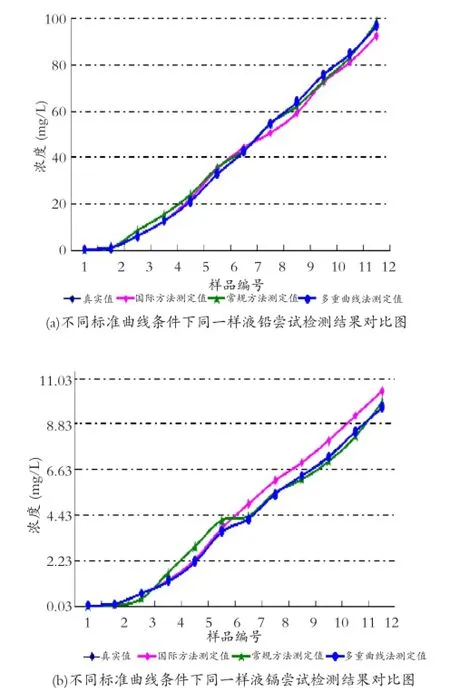
圖2 不同標準曲線條件下12組樣液鉛、鎘濃度檢測結果對比圖Fig.2 The figure of the comparison of testing results of 12 groups of sample liquid under different standard curve condition
4 結 論
(1)受裝飾花面、樣液數量、濃度范圍、儀器設備等因素制約,本文有關不同標準曲線條件對日用陶瓷樣品釉面鉛、鎘溶出量檢測結果準確性影響的結論雖然在理論上符合Lanbert-Beer定律和相關數學原理,對業內實驗室具有普遍的參考價值和現實意義。但針對某未知濃度陶瓷重金屬萃取溶液而言,能否采用多重標準曲線法測定仍需根據具體情況科學對待。
(2)實驗證明在國標法、常規法兩種不同測定條件下,高、中、低各濃度范圍的日用陶瓷重金屬萃取液的測試誤差各異。當樣液中鉛、鎘含量低于40.0 mg/L和4.00 mg/L時,常規方法由于標準曲線的有效覆蓋率有限而存在較大測量誤差,此時國標法不失為一種行之有效的方式。但實施中、高濃度萃取液中鉛、鎘元素測定時,常規方法則以較小的測量不確定度優于國標。
(3)鑒于標準溶液系列與樣品濃度范圍的匹配性對原子吸收光譜法測定日用陶瓷鉛、鎘溶出量的特定要求,應首先利用國標法或常規法估算待測樣液的近似濃度,并據此選取與其最接近的標準溶液系列。從一系列相關的線性擬合回歸方程中,篩選能夠使|A0-?0|最小的方程并以此為基礎繪制標準曲線,可在標準曲線覆蓋的濃度范圍內獲得最小的測量不確定度。
(4)本文依據最小二乘擬合法原理揭示出不同標準曲線條件下,日用陶瓷鉛、鎘溶出量測量不確定度的大小及形成原因,但受測試儀器和樣品數量、樣液濃度等條件所限,實驗分析數據的充分性和代表性受限。因此,針對不同濃度重金屬萃取樣液的檢測需求,如何在一定的測量不確定度條件下,科學設定標準曲線的濃度范圍、標準點數及分布情況,仍有待廣大科技工作者做進一步的研究和探討。
(5)由實驗條件變化所引起的吸光度A的數值變動不可避免,即使在嚴格一致的條件下進行測定時其值也將隨機波動,故吸光度A的測定是一種動態測量。從統計學角度分析原子吸收光譜分析定量測試的基本關系式A=KC并非數學意義上嚴格的函數關系,僅代表吸光度A與濃度C之間的線性相關關系。因此,本文論述的多重標準曲線分析法為臨界超標及仲裁樣品檢測質量控制提供了有益的理論借鑒和實踐參考。
參考文獻:
[1] 袁春梅. 淺談玻璃陶瓷制品鉛、鎘溶出量與人體健康的關系[J]. 輕工標準與質量, 2005, (06): 30-31.
[2] 胡俊, 區卓琨. 日用陶瓷鉛鎘溶出量的成因、危害及檢測[J].佛山陶瓷, 2011, (11): 18-20.
[3] LEHMAN R L. Lead glazes for ceramic food ware [R]. NC USA: The International Lead Management Center, 2002.
[4] PAPANIKOLAOU N C, HATZIDAKI E G, BELIVANIS S, et al. Lead toxicity update: A brief review [J]. Medical Science Monitor, 2005, 11(10): 329-336.
[5] GODT J, SCHEIDIG F, GROSSE-SIESTRUP C, et al. The toxicity of cadmium and resulting hazards for human health [J]. Journal of Occupational Medicine and Toxicology, 2006, 1(22): 1-6.
[6]California Prop. 65-2002 [S].
[7]GB/T 3534-2002《日用陶瓷器鉛、鎘溶出量測定方法》[S].
[8] ASTM C 738-94(2011)《陶瓷制品釉面萃取液中鉛和鎘的標準分析方法》[S].
[9] 劉桂英. 原子吸收光譜法測定重金屬時的干擾因素及消除方法[J]. 中國水利, 2013, (增刊): 77-78.
[10] 喬云峰, 王全九, 莫淑紅等.應用原子吸收分光光度計量測較高濃度鉀溶液[J]. 中國農村水利水電, 2001, (06): 25-27.
[11] 殷海青. 光度分析中工作曲線偏離朗伯一比爾定律的原因[J]. 青海師專學報(教育科學), 2004, (05): 63-66.
[12] 張利興. 多重標準曲線技術在原子吸收法測定鋰同位素豐度中的應用[J]. 計算機與應用化學, 1989, (03): 205-210.
The Application of Multiple Standard Curve Technology in Lead and Cadmium Dissolution Test of Domestic Ceramics by Atomic Absorption Spectrometry
LI Wenjie, DONG Yijun, WANG Haitao
(Ceramic Testing Lab of Tangshan Entry-exit Inspection & Quarantine Bureau, Tangshan 063000, Hebei, China)
This paper summarized the principles of lead and cadmium dissolution measurement of domestic ceramics by atomic absorption spectrometry and the multiple standard curve method. The minimized |A0-?0| was obtained from a series of linear fitting regression equations as the condition of multiple standard curve method for different lead and cadmium concentration ranges of 12 groups of standard sample liquids. The difference between the heavy metal concentration testing results of the same sample liquid obtained from the working curves by the national standard method, the conventional method and the multiple standard method was studied and the cause was analyzed. It is deduced that the accuracy of testing results is higher by national standard method when the concentrations of lead and cadmium are respectively lower than 40.0 mg/L and 4.00 mg/L, but the conclusion is opposite when their concentrations are beyond this scope; the testing error is bigger when the concentrations of lead and cadmium of the sample liquid are respectively between 0.0 mg/L ~ 1.0 mg/L and 0.00 mg/ L ~ 0.10 mg/L by conventional method, but the conclusion is opposite for the medium and high concentration of sample liquid; The degree of uncertainty by multiple standard curve method is the smallest for the entire concentration range of sample liquid.
the multiple standard curve method; atomic absorption spectrometry; measurement; domestic ceramics; the dissolubility of lead and cadmium
TQ174.73
A
1006-2874(2016)05-0043-08
10.13958/j.cnki.ztcg.2016.05.009
2016-06-01。
2016-06-06。
2015年度國家質檢總局科技計劃項目資助(2015IK107)。
李文杰,女,工程師。
Received date:2016-06-01. Revised date: 2016-06-06.
Correspondent author:LI Wenjie, female, Engineer.
E-mail:ribendizhen311@163.com
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
當代陜西(2019年8期)2019-05-09 02:22:48
上海建材(2019年1期)2019-04-25 06:30:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
專用汽車(2016年4期)2016-03-01 04:13:43
質量與標準化(2015年9期)2015-12-31 11:41:40
中國質量與標準導報(2014年4期)2014-03-11 19:54:25
中國質量與標準導報(2014年10期)2014-02-28 22:25:47
中國質量與標準導報(2014年7期)2014-02-28 22:24:39
