環酯的開環聚合
——催化體系與原理
2016-12-17 08:00:13陳永福諸江徽張開炳
山西化工 2016年5期
關鍵詞:催化劑
黎 廣,陳永福,諸江徽,張開炳
(江蘇紅太陽新材料有限公司,江蘇 南京 210000)
?
環酯的開環聚合
——催化體系與原理
黎 廣,陳永福,諸江徽,張開炳
(江蘇紅太陽新材料有限公司,江蘇 南京 210000)
所述環酯泛指具有內酯(lactone)特征的環狀單體,即單體中包括酯基結構。利用不同催化/引發體系,實現環酯的開環聚合制備的生物降解聚酯,具有可循環、可降解等特性,可解決塑料應用的白色污染問題,在生物醫藥、組織工程等領域均有巨大應用潛力。以開環聚合機理區分,應用于環酯開環聚合的催化劑主要是遵循配位-插入機理的金屬催化劑,可分為以下類型:1) 通過親核加成或者親電加成而活化單體;2) 活化引發劑以及增長中的聚合物鏈末端,通過活性末端實現對單體的再引發與鏈增長;3) 對單體與引發劑/增長鏈的雙官能團活化。
開環聚合;環酯;生物降解聚酯;催化
1 含金屬催化劑
金屬催化劑主要可以分為2類:一類是以陰離子催化機理為主的堿金屬催化劑;另一類是以配位-插入機理為主的Sn[1-2]、Al[3]、稀土元素[4-5]、Ca[6]、Mg[7]、Zn[8-9]、Fe[10]等金屬催化劑。其中,堿金屬催化劑主要包括堿金屬的烷基或烷氧基化合物,如,Sec-BuLi[11]、PEG-Li[12-13]、Sec-BuOK[14]等。
在環酯的聚合進展中,符合配位-插入機理的錫(Sn)類金屬鹽對開環聚合制備聚酯的產業化具有巨大的影響。德國的Kricheldorf等對錫類催化機理作出了深入的研究,涉及錫類催化劑對多種環酯單體的作用機制,包括丙交酯、己內酯、三亞甲基碳酸酯等。Kricheldorf等[15]認為,錫類金屬鹽的催化活性來自于金屬中心空sp3d2軌道:在二丁基二異辛酸錫(Bu2SnOct2)中,金屬中心有2個空sp3d2軌道,辛酸基團作為配位基,反應活性受位阻效應而減弱;而異辛酸亞錫(SnOct2)具有3個空sp3d2軌道,路易斯酸性更強,催化活性更高,更適合應用于產業化生產。Kricheldorf[16]還修正了Nijenhuis等對錫類金屬提出的陽離子聚合機理,通過詳細的核磁實驗驗證了金屬中心與單體羰基的配位,單體逐次插入到金屬中心與酸根的共價鍵,此過程既不是陽離子聚合,也不是陰離子聚合,而被定義為 “配位-插入機理”或者“第二軌道插入機理”,見圖1。
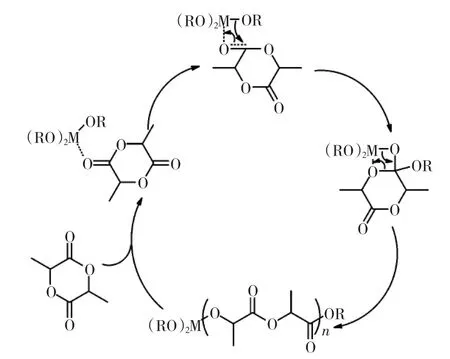
圖1 三價金屬醇鹽的配位-插入機理
然而,隨著研究的深入,人們發現,利用金屬催化得到的聚酯中有部分金屬殘留,可能會導致細胞毒性;殘留金屬在高溫下可恢復催化活性,引起聚合物降解,影響制品質量。當然,以上不足絲毫不會抹殺金屬催化劑在環酯開環聚合中的歷史地位。非金屬有機催化劑的興起則讓聚合物化學更進一層。
20世紀80年代末,Penczek等率先將BF3及HPF6用于催化環醚的開環聚合反應。Hedrick與Waymouth等于2001年報道了DMAP催化環酯的開環聚合,開創了非金屬有機催化劑催化環酯聚合。Hedrick等于2012年獲得美國總統綠色化學獎,以表彰“其研發的有機催化技術避免塑料生產過程中的有害金屬,生產出更安全的產品”。
2 親核加成活化單體的催化劑
此類催化劑多為具有強親核能力的路易斯堿,催化劑親核中心首先進攻環酯的羰基碳原子,形成開環活性中間體。此中間體是一種兼性離子[17],環上酯氧原子帶負電荷。當小分子醇作為引發劑時,醇羥基氫被酯氧負離子奪取,生成的羥基氧負離子親核加成到開環單體的羰基碳上,同時釋放催化劑,以活化下一個單體。鏈增長中的中間體取代原小分子醇,接受單體插入到羥基氫、氧原子之間實現鏈增長。這類催化劑以DMAP與N-雜環卡賓(NHCs)為代表。
DMAP分子中的取代二甲胺基與雜環形成共振,能有效增強環上氮原子的孤對電子的親核性。多種小分子醇可被用作開環引發劑,最終產物具有羥基末端。除環酯單體,Bourissou等還以DMAP催化了五元環O-內羧酸酐的開環聚合,通過反應路徑能量計算,證實了DMAP作為親核試劑可促進內羧酸酐的脫羧開環過程。Brignou和Helou分別報道了DMAP催化六元環狀碳酸酯的本體聚合,但所得產物相對分子質量分布較寬。
另一種應用在開環聚合中的經典親核催化劑為N-雜環卡賓,如圖2所示。在醇/NHCs形式的引發/催化體系中,環酯的開環聚合在溫和條件下反應速度快,且體現活性聚合的特征。NHCs催化開環聚合反應機理與DMAP相同,利用NHCs的親核性活化單體,從而達到開環目的,如圖3所示。2002年,Hedrick等首次報道了NHC催化丙交酯的開環聚合反應,其單體活化的機理還通過制備大環聚合物的實驗得到了進一步驗證。無引發劑時,NHC對單體親核進攻后形成的兼性離子將以末端酯氧負離子進一步引發單體開環。當單體轉化完全時,氧負離子將對NHC的親核位點再一次進攻,游離出NHC,并得到大環聚合物[21]。
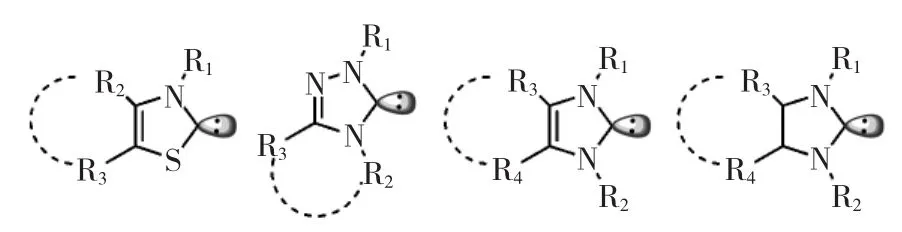
圖2 N-雜環卡賓
2009年,Hedrick等將NHCs引入ε-CL聚合中,當N原子取代基為芳香族基團時,反應極緩慢,甚至不進行;當N取代基為短鏈烷基時,反應較為順利。這說明,NHC作為親核試劑,N原子取代基對于親核位點的暴露程度有較大立體空間影響。NHC體系是膦催化向碳催化發展的奠基石[18]。
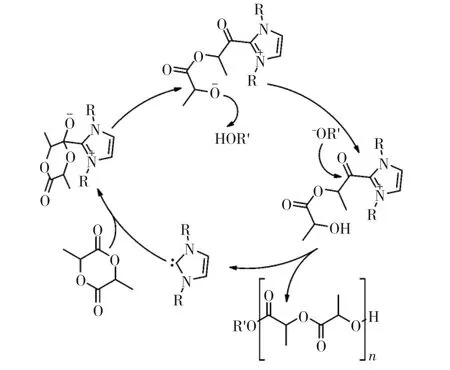
圖3 NHC催化丙交酯開環聚合機理
3 親電加成活化單體的催化劑
此類催化劑與傳統陽離子催化劑相似。Penczek等認為,氧鎓離子的正電荷最終由碳原子承載,形成具有親電加成能力的碳正離子。碳正離子的親電加成過程構成了鏈的增長。此過程活化對象始終為單體,電荷轉移使其他單體親電加成到聚合物鏈末端的電負性中心。一般認為,布朗斯特酸/醇體系催化環酯的開環聚合機理屬于陽離子活化單體機理(見圖4)。質子氫首先活化羰基氧,使得羰基碳的正電性加強;之后,醇上的羥基氧原子對環上的羰基碳進行親核進攻,引發開環。
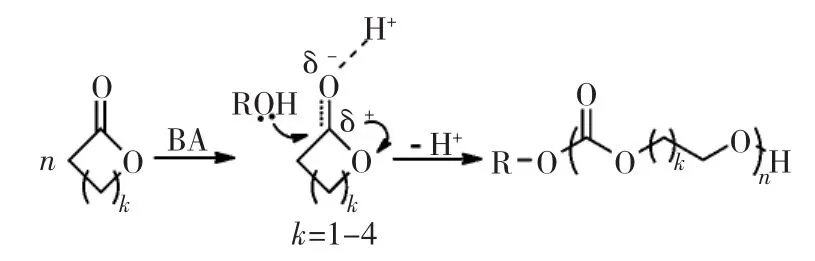
圖4 布朗斯特酸/醇體系催化內酯開環聚合
Kricheldorf等在20世紀80年代對酸催化環酯開環聚合進行了廣泛研究。以單體轉化率和產物聚合度為依據,他們認為,只有磺酸及其衍生物才是環酯開環聚合的有效催化劑。Bourissou等以甲基磺酸(CH3SO3H,MSA)催化了丙交酯的開環聚合,MALDI-TOF MS數據證明,磺酸類催化丙交酯依據陽離子單體活化機理,不會發生烷氧鍵斷裂。Bourissou等還研究了MSA與三氟甲基磺酸(CF3SO3H,TfOH)催化TMC的開環聚合,發現MSA在催化TMC開環聚合中得到的聚合產物PDI低。
近5年來,全氟代磺酸憑借其高催化活性得到關注。2009年,Kakuchi等首次將超強布朗斯特酸雙三氟磺酰亞胺((CF3SO2)2NH,HNTf2)用于甲基丙烯酸酯的基團轉移聚合反應。該課題組于2010年以HNTf2成功地催化了δ-戊內酯(δ-VL)的開環聚合反應,反應以苯丙醇為引發劑,二氯甲烷為溶劑,聚合過程可控且表現出活性聚合的特征。2011年,Oshimura等報道了范圍更廣的全氟代磺酸催化ε-CL開環聚合反應,室溫下在甲苯中反應8 h,獲得的PCL的相對分子質量2 000 g/mol~6 400 g/mol,PDI=1.10~1.48。全氟代磺酸具有超強的酸性,甚至在以四氫呋喃為溶液中的開環聚合反應中,含有聚四氫呋喃片段。圖5為用于開環聚合的有機酸類催化劑。
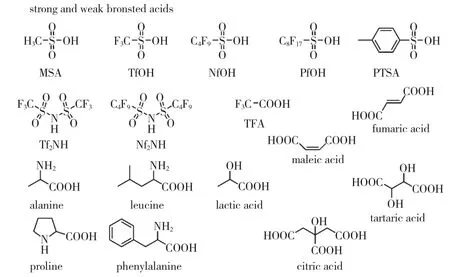
圖5 用于開環聚合的有機酸類催化劑
除了上述強酸與超強酸外,弱酸,如三氟乙酸(CF3CO2H,TFA)[19]、天然α-羥基酸和α-氨基酸[20-22],同樣可用于環酯的開環聚合中。
4 鏈端活化催化劑
此類催化劑一般為布朗斯特堿,體系中還包括小分子醇作為引發劑。催化劑首先奪取羥基氫,形成具有親核進攻/引發能力的氧負離子。親核/引發中心始終位于鏈末端,這與傳統陰離子開環聚合機理相似,是理論上的活性聚合。活化單體機理與活化鏈端機理所涉及的催化劑存在一定交叉,不存在也沒有必要進行絕對區分。例如,低溫下,經典的活化單體類型催化劑NHC催化消旋丙交酯開環聚合所得產物具有高度立構選擇性[23],立構度的控制體現了末端活化機理。NHC作為布朗斯特堿奪取醇羥基質子,氧負離子作為親核試劑進攻單體。
除了生成陰離子活性中心的引發形式,醇和親核試劑間的氫鍵也被認為可實現鏈端活化機理。以DBU為例,當存在小分子醇作為引發劑時,DBU首先與羥基形成氫鍵[24-25],增強羥基氧的親核能力。開環產物始終保持末端為羥基,DBU進而活化開環產物末端,以提高末端親核能力,開環下一個單體。值得注意的是,當不存在小分子引發劑時[26],DBU作為親核試劑直接進攻單體羰基引發開環。催化特點與NHC等親核加成/單體活化催化劑完全一致,開環中間體亦為兼性離子,產物為大環聚合物,這體現了DBU的單體活化能力。圖6為用于開環聚合的胍類與脒類催化劑。
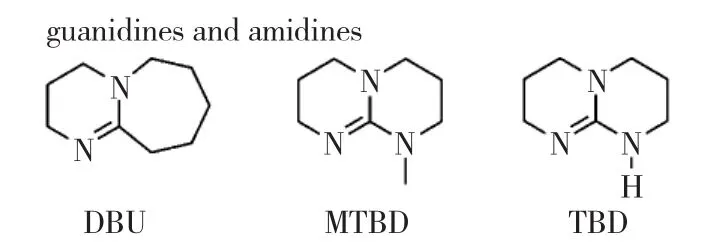
圖6 用于開環聚合的胍類與脒類催化劑
另一種代表性催化劑為磷腈類化合物。例如,2-叔-丁基亞氨基-2-二乙胺基-1,3-二甲基全氫-1,3,2-二吖磷英(2-tert-butylimino-2-diethylamino-1,3-dimethyl-perhydro-1,3,2-diazaphosphorine,BEMP)催化環酯開環聚合反應機理如第49頁圖7。首先,通過氫鍵作用活化引發劑/鏈端,形成親核試劑引發開環,活性中心始終為鏈末端的氧負離子。2007年,Hedrick等首先報道了磷腈催化丙交酯的開環聚合,并以核磁滴定的實驗證明了磷腈對引發劑醇的末端活化。在環酯的開環聚合中,磷腈展現了極高的催化活性,在低溫下(-75 ℃)反應3 h,單體轉化率>99%。此外,二聚膦腈還擁有高度立體選擇性,催化內消旋丙交酯可得全同立構聚合物。
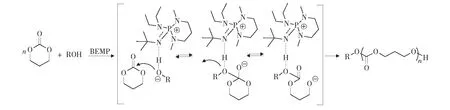
圖7 BEMP催化TMC開環聚合機理
5 雙官能團催化劑
有機化學中不對稱反應對高選擇性催化劑的需求,促進了雙官能團催化劑的設計與發展[27-28]。作為制備富含對映體化合物的重要手段,其傳統催化劑研究方向為在過渡金屬中心構建手性位點[29-30]。而純有機催化劑開發理念為:1) 反應活性較弱的親電中心通過與手性布朗斯特/路易斯酸的配位(或稱之為協同締合)作用,反應活性增強,更易于親核加成得到富含對映體的產物;2) 反應活性較弱的親核中心通過與手性布朗斯特/路易斯堿的配位作用,親核能力得到增強,與親電中心加成后得到光學活性化合物。然而,在眾多反應中,底物的反應活性較弱,難以有效生成所需要的對映體。所以,在近20年中,同時活化親核中心與親電中心的雙官能團催化劑成為研究熱點[31-33]。
雙官能團催化劑典型的模型就是,一端包含1個酸性位點,可以與底物親電基團產生締合活化作用;而另一端則包含1個堿性位點,可以活化親核試劑。雙官能團催化劑的2種催化中心,一般所處的化學環境相似,并具有與設計相符的構象。雙官能團催化劑可以協同促進反應,這與酶催化機理類似。在環酯的開環聚合反應中,成熟的雙官能團催化劑主要包括硫脲-胺體系、TBD、噻唑-胺體系。
2005年,Hedrick等首次將硫脲-胺體系引入到環酯的開環聚合中,催化了L-丙交酯的開環聚合,見圖8。從核磁結果中觀察到的聚合物立規度與產物熔點可知,硫脲-胺體系并不會對丙交酯的次甲基手性位點造成消旋。反應體系于48 h內無轉酯反應,反應過程中相對分子質量分布一直控制在較低水平,呈現活性聚合特征。隨后,更多形式的硫脲-胺的體系被引入開環聚合反應中[34-35],無論是修飾胺與硫脲的連接位點,還是改變硫脲的取代基團,亦或直接混合單獨的硫脲和胺,都可以體現對開環聚合反應的催化活性與聚合控制。
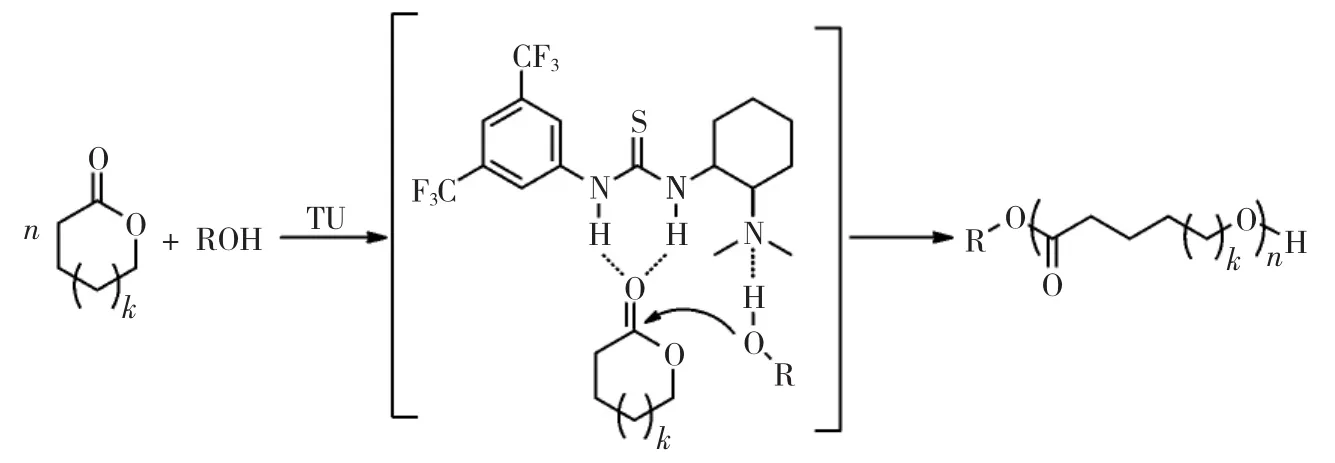
圖8 硫脲-胺作為雙官能團催化劑催化內酯開環聚合
對于ε-CL和δ-VL體系,一般的堿難以單獨實現對其開環聚合,即便是強親核試劑,如N-雜環卡賓,也需要降低N原子取代基的體積,以暴露親核活性中心。鑒于較為常用的胺類堿DBU、MTBD不能獨立催化ε-CL和δ-VL的開環聚合,Hedrick等向體系中添加了與堿濃度一致的硫脲,可得到數均相對分子質量過萬的聚合物,體現了硫脲與堿的協同作用。在四氫呋喃和二甲基甲酰胺等含有氫鍵的溶劑中,反應無產物生成。這從側面說明,硫脲對單體羰基氧原子通過形成氫鍵進行活化,受溶劑效應影響較大。
硫脲-胺催化內酯開環聚合的反應機理總結如下:內酯單體的羰基氧與硫脲形成強氫鍵,羰基雙鍵電子分布偏向氫鍵締合位點,增強了羰基碳原子的親電性;同時,作為引發劑的醇或者增長中聚合物的羥基氫與叔胺形成氫鍵,羥基氧的親核性得到增強,更易親核加成到單體羰基碳原子上引發開環過程。熱力學上,隨著單體環張力被釋放,聚合反應得到推動。即使延長反應時間至48 h~72 h,在單體完全消耗的情況下,體系中極少存在轉酯反應,反應呈活性聚合特征。
在Hedrick的工作中,以TBD為催化劑催化了L-LA、ε-CL、δ-VL、β-BL的開環聚合。通過TBD與單體1/1混合樣品的核磁分析得出結論,TBD作為質子梭在體系中轉移仲胺質子至單體以及聚合物末端。在仲胺給出質子、亞肟親核進攻羰基碳后,初始仲胺雜氮原子化軌道變為sp2,形成雙鍵,形成新的亞肟結構;而初始亞肟氮原子與羰基碳形成穩定N-C鍵,為sp3雜化軌道,形成叔胺。新形成的亞肟氮原子對開環產物末端羥基氫有活化作用,聚合過程依靠末端羥基氧原子對下一個單體親核進攻。而亞肟氮原子對醇末端氫的活化也通過核磁滴定實驗進行了驗證,羥基氫在形成締合之后,化學位移向低場有明顯偏移,峰形尖銳。Goodman等[36]以密度泛函理論計算,演示并驗證了TBD對于環酯單體的活化形式。TBD作為雙官能團催化劑催化酯的氨解反應見圖9。

圖9 TBD作為雙官能團催化劑催化酯的氨解反應
6 總結
從增長形式上來說,開環聚合過程中的鏈增長類似于鏈式聚合,即單體逐一接入聚合物鏈的活性末端。但從單體的形式上來說,開環聚合更為復雜,因為不同的單體活化形式決定了反應速率與聚合控制程度。而開環聚合成為研究熱點的最主要原因在于,可以高效可控地合成出科學前沿新型聚合物。通過對環狀單體的修飾,使聚合物擁有特殊性質(例如,環境響應性、指定范圍的折光系數)。開環聚合反應適用于制備天然聚合物(例如,聚多糖、聚氨基酸等)與生物降解聚合物(例如,聚乳酸、聚己內酯等)。各催化劑及引發體系各有特點與利弊,并在不同單體反應中體現出不同作用。
[1] Kricheldorf H,Stricker A.Polymers of carbonic acid 29.Bu2SnOct2-initiated polymerizations of trimethylene carbonate (TMC,1,3-dioxanone-2)[J].Polymer,2000,41(20):7311-7320.
[2] Bhaw-Luximon A,Jhurry D,Motala-Timol S,et al.Polymerization of ε-caprolactone and its copolymerization with γ-butyrolactone using metal complexes:proceedings of the macromolecular symposia[C].Wiley Online Library,2005.
[3] Koller,Bergman R G.Highly efficient aluminum-catalyzed ring-opening polymerization of cyclic carbonates,lactones,and lactides,including a unique crystallographic snapshot of an intermediate[J].Organometallics,2011,30(11):3217-3224.
[4] Shen Y,Shen Z,Zhang Y,et al.A comparison of polymerization characteristics and mechanisms of ε-caprolactone and trimethylene-carbonate with rare earth halides[J].Journal of Polymer Science:Part A:Polymer Chemistry,1997,35(8):1339-1352.
[5] Takeuchi D,Nakamura T,Aida T.Bulky titanium bis (phenolate) complexes as novel initiators for living anionic polymerization of ε-caprolactone[J].Macromolecules,2000,33(3):725-729.
[6] Zhong Z,Schneiderbauer S,Dijkstra P J,et al.Single-site calcium initiators for the controlled ring-opening polymerization of lactides and lactones[J].Polymer Bulletin,2003,51(3):175-182.
[7] Darensbourg D J,Choi W,Ganguly P,et al.Biometal derivatives as catalysts for the ring-opening polymerization of trimethylene carbonate:Optimization of the Ca(Ⅱ) salen catalyst system[J].Macromolecules,2006,39(13):4374-4379.
[8] Chamberlain B M,Cheng M,Moore D R,et al.Polymerization of lactide with zinc and magnesium β-diiminate complexes:Stereocontrol and mechanism[J].Journal of the American Chemical Society,2001,123(14):3229-3238.
[9] Cheng M,Attygalle A B,Lobkovsky E B,et al.Single-site catalysts for ring-opening polymerization:Synthesis of heterotactic poly (lactic acid) from rac-lactide[J].Journal of the American Chemical Society,1999,121(49):11583-11584.
[10]Dobrzynski P,Pastusiak M,Bero M.Less toxic acetylacetonates as initiators of trimethylene carbonate and 2,2-dimethyltrimethylene carbonate ring opening polymerization[J].Journal of Polymer Science:Part A:Polymer Chemistry,2005,43(9):1913-1922.
[11]Müller A J,Keul H,H?cker H.Lithium and potassium alcoholates of poly (ethylene glycol) s as initiators for the anionic polymerization of 2,2-dimethyltrimethylene carbonate:Synthesis of AB and ABA block copolymers[J].European Polymer Journal,1991,27(12):1323-1330.
[12]Müller A J,Keul H,H?cker H.Synthesis and thermal properties of poly (2,2-dimethyltrimethylene carbonate)-block-poly (tetrahydrofuran)-block-poly (2,2-dimethyltrimethylene carbonate)[J].European Polymer Journal,1993,29(9):1171-1178.
[13]Matsuo J,Aoki K,Sanda F,et al.Substituent effect on the anionic equilibrium polymerization of six-membered cyclic carbonates[J].Macromolecules,1998,31(14):4432-4438.
[14]Kricheldorf H R,Kreiser-Saunders I,Boettcher C.Polylactones:31:Sn(Ⅱ) octoate-initiated polymerization of L-Lactide:A mechanistic study[J].Polymer,1995,36(6):1253-1259.
[15]Kricheldorf H R,Kreiser-Saunders I,Stricker A.Polylactones:48:SnOct2-initiated polymerizations of lactide:A mechanistic study[J].Macromolecules,2000,33(3):702-709.
[16]Kricheldorf H R,Serra A.Polylactones[J].Polymer Bulletin,1985,14(6):497-502.
[17]Albertsson A C,Varma I K.Recent developments in ring opening polymerization of lactones for biomedical applications[J].Biomacromolecules,2003,4(6):1466-1486.
[18]Brignou P,Priebe Gil M,Casagrande O,et al.Polycarbonates derived from green acids:ring-opening polymerization of seven-membered cyclic carbonates[J].Macromolecules,2010,43(19):8007-8017.
[19]Bourissou D,Martin-Vaca B,Dumitrescu A,et al.Controlled cationic polymerization of lactide[J].Macromolecules,2005,38(24):9993-9998.
[20]Delcroix D,Martín-Vaca B,Bourissou D,et al.Ring-opening polymerization of trimethylene carbonate catalyzed by methanesulfonic acid:Activated monomer versus active chain end mechanisms[J].Macromolecules,2010,43(21):8828-8835.
[21]Shibasaki Y,Sanda F,Endo T.Cationic ring-opening polymerization of seven-membered cyclic carbonate with water-hydrogen chloride through activated monomer process[J].Macromolecules,2000,33(10):3590-3593.
[22]Kakuchi R,Tsuji Y,Chiba K,et al.Controlled/living ring-opening polymerization of δ-valerolactone using triflylimide as an efficient cationic organocatalyst[J].Macromolecules,2010,43(17):7090-7094.
[23]Oshimura M,Tang T,Takasu A.Ring-opening polymerization of ε-caprolactone using perfluoroalkanesulfonates and perfluoroalkanesulfonimides as organic catalysts[J].Journal of Polymer Science:Part A:Polymer Chemistry,2011,49(5):1210-1218.
[24]Dove A P,Li H,Pratt R C,et al.Stereoselective polymerization of rac-and meso-lactide catalyzed by sterically encumbered N-heterocyclic carbenes[J].Chemical Communications,2006(27):2881-2883.
[25]Zhang L,Pratt R C,Nederberg F,et al.Acyclic guanidines as organic catalysts for living polymerization of lactide[J].Macromolecules,2010,43(3):1660-1664.
[26]Lohmeijer B G,Pratt R C,Leibfarth F,et al.Guanidine and amidine organocatalysts for ring-opening polymerization of cyclic esters[J].Macromolecules,2006,39(25):8574-8583.
[27]Zhang L,Nederberg F,Messman J M,et al.Organocatalytic stereoselective ring-opening polymerization of lactide with dimeric phosphazene bases[J].Journal of the American Chemical Society,2007,129(42):12610-12611.
[28]Kanai M,Kato N,Ichikawa E,et al.Power of cooperativity:Lewis acid-Lewis base bifunctional asymmetric catalysis[J].Synlett,2005,2005(10):1491-1508.
[29]Paull D H,Abraham C J,Scerba M T,et al.Bifunctional asymmetric catalysis:Cooperative Lewis acid/base systems[J].Accounts of Chemical Research,2008,41(5):655-663.
[30]Noyori R,Yamakawa M,Hashiguchi S.Metal-ligand bifunctional catalysis:A nonclassical mechanism for asymmetric hydrogen transfer between alcohols and carbonyl compounds[J].The Journal of Organic Chemistry,2001,66(24):7931-7944.
[31]Ikariya T,Murata K,Noyori R.Bifunctional transition metal-based molecular catalysts for asymmetric syntheses[J].Organic & Biomolecular Chemistry,2006,4(3):393-406.
[32]Cole A C,Jensen J L,Ntai I,et al.Novel br?nsted acidic ionic liquids and their use as dual solvent-catalysts[J].Journal of the American Chemical Society,2002,124(21):5962-5963.
[33]Inokuma T,Hoashi Y,Takemoto Y.Thiourea-catalyzed asymmetric michael addition of activated methylene compounds to α,β-unsaturated imides:Dual activation of imide by intra-and intermolecular hydrogen bonding[J].Journal of the American Chemical Society,2006,128(29):9413-9419.
[34]Dove A P,Pratt R C,Lohmeijer B G,et al.Thiourea-based bifunctional organocatalysis:Supramolecular recognition for living polymerization[J].Journal of the American Chemical Society,2005,127(40):13798-13799.
[35]Alba A,Schopp A,De Sousa Delgado A P,et al.Controlled ring-opening polymerization of lactide by bis-sulfonamide/amine associations:Cooperative hydrogen-bonding catalysis[J].Journal of Polymer Science:Part A:Polymer Chemistry,2010,48(4):959-965.
[36]Simón L,Goodman J M.The mechanism of TBD-catalyzed ring-opening polymerization of cyclic esters[J].The Journal of Organic Chemistry,2007,72(25):9656-9662.
Ring opening and polymerization of cyclic ester——catalytic system and principles
LI Guang, CHEN Yongfu, ZHU Jianghui, ZHANG Kaibing
(Jiangsu Red Sun New Materials Co., Ltd., Nanjing Jiangsu 210000, China)
Cyclic ester described in this paper is cyclic monomer with characteristics of lactone (lactone) , monomers include the structure of ester base. Using different initiator/catalyst system, it can realize ring opening polymerization of cyclic ester to produce biodegradable polyester, it is renewable, biodegradable, and it can solve the white pollution problems of the application of plastic, and have huge potential application in the field of biological medicine, tissue engineering and so on. Using ring opening polymerization mechanism to distinguish, catalyst applied to ring opening polymerization of cyclic ester is mainly metal catalysts which following coordination-insertion mechanism. It can be divided into the following types: 1) Through nucleophilic addition or electrophilic addition to active monomer. 2) To activatethe initiator and the end of the polymer chain, through active end to realise re-initiation and chain growth of monomer. 3) Toactivate double functional groups of monomer and initiator/growing chain.
ring opening and polymerization; cyclic ester; biodegradable polyester; catalyze
2016-04-25
黎 廣,男,1985年出生,畢業于山西大學,碩士學位。研究方向:工業催化。
綜述與論壇
10.16525/j.cnki.cn14-1109/tq.2016.05.14
O643.3
A
1004-7050(2016)05-0046-07
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
