鰻鱺肌肉中孔雀石綠代謝物隱性孔雀石綠染料殘留標準物質的研制
2016-12-20 00:58:57尹太坤劉正才林永輝
水產科學 2016年3期
尹太坤,楊 方,劉正才 ,林永輝
( 1.福州大學 石油化工學院 福建 福州 350108; 2.福建出入境檢驗檢疫局 檢驗檢疫技術中心,福建 福州 350001 )
鰻鱺肌肉中孔雀石綠代謝物隱性孔雀石綠染料殘留標準物質的研制
尹太坤1,楊 方2,劉正才2,林永輝2
( 1.福州大學 石油化工學院 福建 福州 350108; 2.福建出入境檢驗檢疫局 檢驗檢疫技術中心,福建 福州 350001 )
為建立鰻鱺肌肉中孔雀石綠代謝物隱性孔雀石綠染料殘留標準物質的研制和定值方法,以一定質量濃度孔雀石綠對鰻鱺進行藥浴給藥,使孔雀石綠在魚體內自然代謝,從而使鰻鱺體內含有隱性孔雀石綠殘留。經均質、真空包裝及輻照處理后,獲得一批500個獨立包裝的的鰻鱺肌肉樣本。采用超高效液相色譜-串聯質譜法對該樣本進行均勻性和穩定性檢驗,經8家獨立實驗室協同定值及不確定度評估,其特性值為2.82 μg/kg,擴展不確定度為0.39 μg/kg(k=2)。所建立的制備方法為染料殘留鰻鱺基體標準物質的實驗室制備提供了一種參考。
鰻鱺肌肉;隱性孔雀石綠;殘留;標準物質
標準物質是具有一種或多種足夠均勻和很好地確定了的特性值,用于校準設備、評價測量方法或給材料賦值的材料或物質[1],在保證分析方法的準確性及可溯源性方面具有重要作用[2]。標準物質分為純品標準物質和基體標準物質兩類,后者的候選材料獲取于天然基體,目標物和基體結合情形與真實檢測樣品完全一致,在分析中可有效避免基體效應對物質成分分析的影響。
孔雀石綠屬三苯甲烷類染料,曾作為驅蟲劑和殺菌劑廣泛應用于水產養殖業,其在生物體內會代謝為隱性孔雀石綠[3]。隱性孔雀石綠具有較高的親脂性,在魚類脂肪組織中代謝速度較慢,會形成穩定殘留并有明顯的蓄積現象[4-5]。由于孔雀石綠及其代謝物隱性孔雀石綠具有致癌性、高毒性、高殘留的特點[6-10],美國、日本、歐盟等國家已禁止孔雀石綠在水產品中使用, 我國也于2002年5月將孔雀石綠列入《食品動物禁用的獸藥及其化合物清單》 中嚴禁使用。因此,孔雀石綠及其代謝物隱性孔雀石綠已成為殘留檢測實驗室日常檢測項目,開展食品中殘留監控具有重要意義。
近年來有報道關于含藥物殘留的動物源基體標準物質的研制[11-15],但尚未見含孔雀石綠代謝物隱性孔雀石綠染料殘留的鰻鱺(Anguillajaponica)肌肉基體標準物質。本文介紹了以鰻鱺肌肉為基體的含孔雀石綠代謝物隱性孔雀石綠的標準物質的研制過程,該標準物質已在實際工作中得到應用,用于檢測結果的質量控制。
1 材料與方法
1.1 儀器與試劑
Waters UPLC/Premier超高效液相色譜-串聯質譜儀,配有電噴霧離子源(美國Waters公司);直線電子加速器(同方威視IS075型,7.5 MeV,5 kW);勻漿機(丹麥FOSS公司);拍擊均質器(英國Seward公司);Bond Elut 氧化鋁SPE固相萃取柱(中性,安捷倫科技有限公司)。
隱性孔雀石綠標準品(純度≥98.5%)和隱性孔雀石綠-D6氚代同位素內標(純度≥99.8%),均購于德國 Dr.Ehremstorfer公司;乙腈為色譜純,購于德國Merck公司;包裝材料:鋁箔復合材料平口袋為外包裝,聚乙烯平口袋作內袋。
1.2 自然污染樣本的制備
選取約250 g已確認不含目標物的鰻鱺,于養殖箱中養殖,水溫20~22 ℃,采用空氣泵24 h增氧。配制質量濃度約為0.03 mg/L的孔雀石綠溶液,以全池噴灑方式給藥,用藥3 d后采集陽性樣本,均質混勻,制成肉糜。采用雙層復合袋獨立真空包裝,每袋約15 g,粘貼樣品標簽,獲得獨立包裝樣本500個。
1.3 樣品均勻性與穩定性檢驗
隨機抽取15份獨立包裝樣品,取樣量為2 g,以每個樣品3次重復測試數據為一組,每種藥物共15組45個數據,采用單因素方差分析法(F-檢驗)檢驗樣品的均勻性。
穩定性檢驗分為長期穩定性和短期穩定性進行,長期穩定性共進行7個月,每月取樣1次,每個樣品重復檢測3次,以平均值作為檢測結果。短期穩定性通過模擬樣品運輸條件,利用F-檢驗法對檢測結果的均勻性進行評價。
1.4 定值及溯源性
本批標準品由8家認可實驗室為本殘留質量控制樣品協同定值,檢測數據經夏皮洛—威爾克檢驗[16]、柯克倫檢驗及格拉布斯檢驗[17]后取平均值進行定值。特性值不確定度UCRM參照《標準樣品工作導則(3)標準樣品定值的一般原則和統計方法》[18]進行評估。
標準物質在空間上具有溯源量值的功能[19]。本研究選擇的溯源路線是,用有證標準物質內標法定量確定含量,通過有證標準物質溯源到基準單位[20]。以有證標準物質配制的隱形孔雀石綠標準溶液制作校準曲線;測量前對所有計量儀器(分析天平、容量瓶、移液槍等)進行了校準,考慮其示值不確定度;分析方法采用《水產品中孔雀石綠和結晶紫殘留量的測定》[21];具有資質的8家實驗室根據檢測情況選用不同的已被證實的檢測方法定值等,保證了結果的準確性和溯源性[22]。
1.5 樣品的分析方法
按《水產品中孔雀石綠和結晶紫殘留量的測定》[21]檢測樣品中隱性孔雀石綠殘留量。采用乙腈超聲提取,經中性氧化鋁柱凈化后,超高效液相色譜-串聯質譜法測定,同位素內標法定量。檢測離子對m/z:331.3/315.7(定量)、331.3/239.5(定性)、337.3/240.2(LMG-D6)。
2 結果與分析
2.1 鰻鱺體內藥物代謝
進行鰻鱺給藥試驗,連續藥浴2 d后停用藥物,清洗水池,換100%清水(不含藥物)養殖。經取樣檢測,鰻鱺肌肉中隱性孔雀石綠的殘留量隨藥后時間的變化趨勢見圖1。隨時間的推移,血液和鰻鱺肌肉中隱性孔雀石綠的殘留量均先升后降。空白樣本于施藥前采集,陽性樣本的采集時間考慮到預期質量濃度范圍,選擇藥后第3 d采集自然污染樣本。將空白樣本與陽性樣本分別宰殺,去頭、放血、除去內臟和魚皮,置于冰柜-18 ℃冷凍保存。
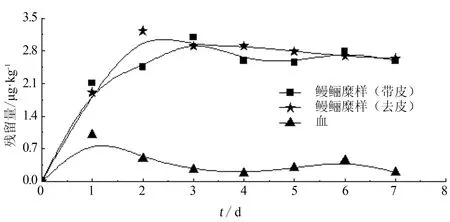
圖1 鰻鱺肌肉中隱性孔雀石綠殘留量隨藥后時間變化
2.2 樣本的制備
將鰻鱺肌肉切段(每段長約2 cm)置于勻漿機中,3000 r/min 轉速攪成糜樣并勻漿5 min;以4 kg/次的鰻鱺肌肉量攪拌3次,混合后再于大桶中人工攪拌20 min;再采用拍擊式均質器進行二次混勻:將樣品裝入拍擊袋,置于拍擊式均質器中,按180拍/min的頻率,每次拍擊5 min后取出,再進行更換拍擊,如此反復拍擊3次。對陽性肌肉樣本,采用搗碎勻漿、拍擊均質和人工攪拌相結合的工藝,保障了樣本的均勻性。選用聚乙烯平口袋為內袋,鋁箔復合材料為外袋的雙層包裝袋進行真空包裝后,對樣本進行60Co-γ輻照滅菌,有效延長了保質期。輻照前后樣本中隱性孔雀石綠殘留量見表1,結果表明經60Co-γ輻照后,鰻鱺基體中隱性孔雀石綠基本不降解,輻照后目標物含量仍處于預期質量濃度范圍內。
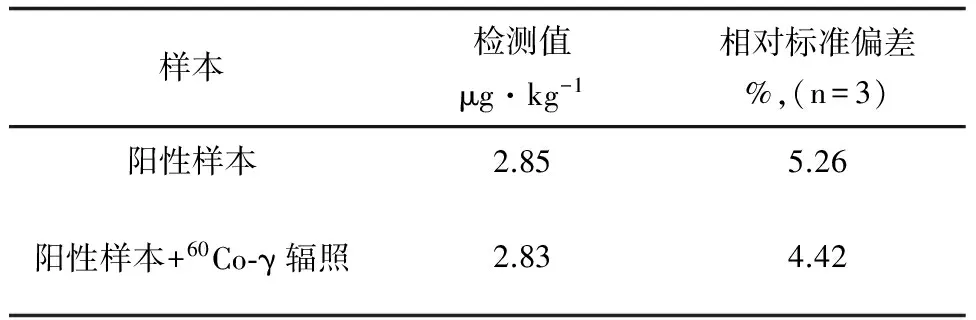
表1 60Co-γ輻照對鰻鱺肌肉中隱性孔雀石綠殘留量的影響
2.3 樣品譜圖
0.5 μg/kg 隱性孔雀石綠標準溶液的色譜圖見圖2a;陽性樣本按1.5前處理后上機檢測,得到離子監測色譜圖(樣本含量2.91 μg/kg)見圖2b。由圖2可見,各離子對的峰形良好,且目標物出峰處無雜峰干擾,方法檢出限為0.5 μg/kg,由此可見,制備的鰻鱺肌肉中隱性孔雀石綠的質量濃度適當。
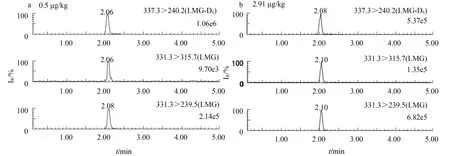
圖2 標準溶液(a) 及樣品(b)的選擇離子監測色譜圖
2.4 均勻性檢驗
隨機抽取15份獨立包裝樣品,每個樣品平行檢測3次。以每袋含量的平均值作為對袋序號的函數來研究樣品制備中的趨勢和分裝過程的邏輯關系(圖3)。由此可見樣品制備中的趨勢穩定;分裝均一,數據穩定。
對檢測結果采用單因素方差分析法(F-檢驗)進行統計分析,方差分析結果見表2。F值=1.182
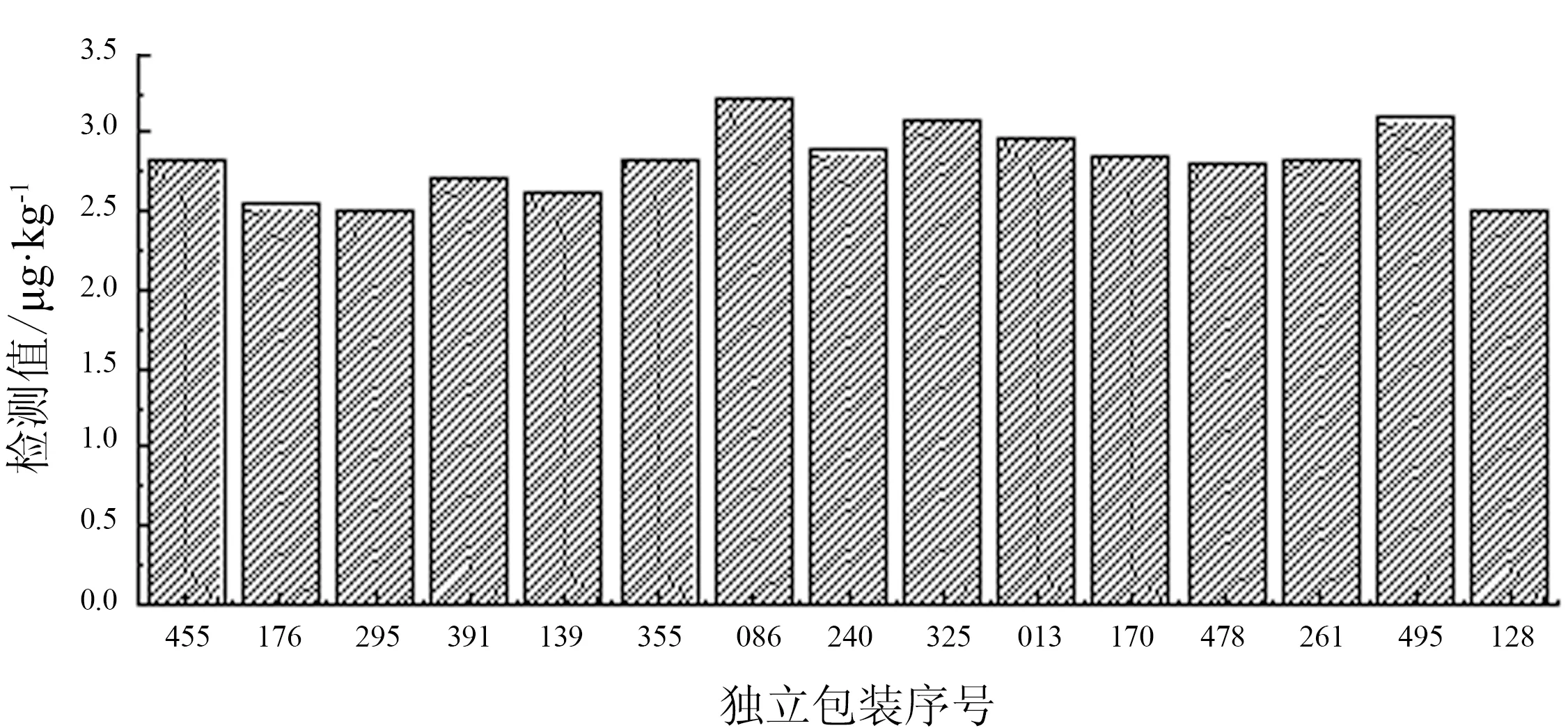
圖3 樣本均勻性檢驗數據平均值
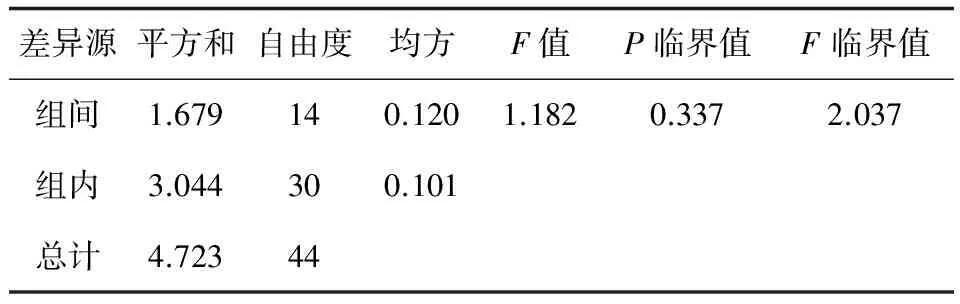
表2 鰻鱺肌肉中隱形孔雀石綠殘留量的均勻性方差分析
2.5 穩定性檢驗
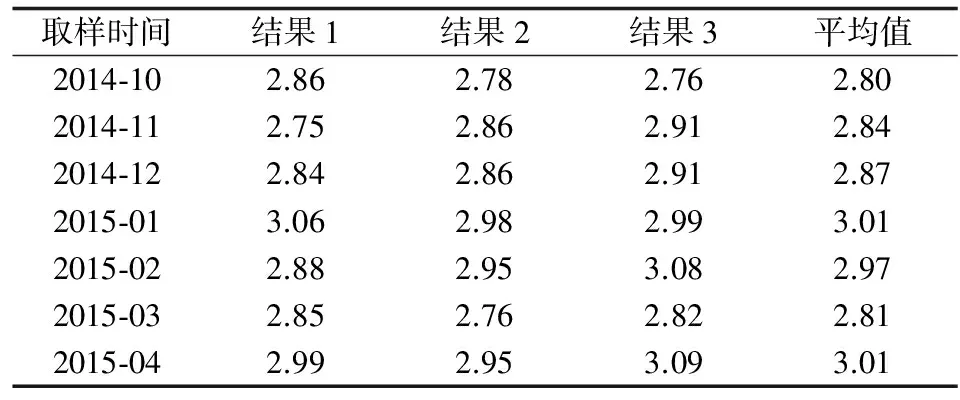
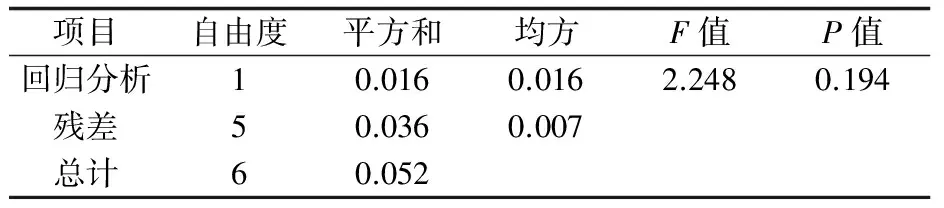
2.5.1 短期穩定性
短期穩定性試驗模擬樣品運輸條件,將樣本分別置于-70 ℃、4 ℃、室溫、陽光下曝曬條件下保存14 d后測定,考察短周期穩定性。每個樣品重復測試3次,采用單因素方差分析法,分析結果見表3。在顯著水平α=0.05時,F=0.501 表3 短期穩定性方差分析 2.5.2 長期穩定性 《標準樣品工作導則(3)標準樣品定值的一般原則和統計方法》[18]提供了2種檢驗方法,余孔捷等[12,14-15]采用F檢驗法檢驗一元線性回歸模型的顯著性,通過評估回歸方差,檢驗MSreg與s2比值的顯著性,對于95%的置信水平,當P≥0.05時,表示回歸不顯著,判定為樣品穩定。楊麗君等[23]采用t檢驗法,通過誤差分析計算回歸參數b1(斜率)的標準偏差s(b1),當滿足|b1| 表4 隱性孔雀石綠長期穩定性測定結果(n=3) μg/kg 表5 長期穩定性方差分析 本批標準品采用多實驗室協同定值的方案,大連出入境檢驗檢疫局、浙江出入境檢驗檢疫局、河北出入境檢驗檢疫局、廣西出入境檢驗檢疫局、廈門出入境檢驗檢疫局、深圳出入境檢驗檢疫局、福建中檢華日有限公司、福建省產品質量檢驗研究院8家具有檢測資質的機構共同定值,分別用1~8編號表示。雖檢測環境、儀器條件、檢測人員不同,但均采用液相色譜-串聯質譜法進行測定(n=3),每個實驗室檢測結果見表6。 表6 協同定值 μg/kg 為保準后續定值的準確性,需對8家實驗室的檢測值進行離群值檢驗,剔除無效數據。楊方等[11-12,15]在定值前對各實驗室的檢測值進行了柯克倫檢驗及格拉布斯檢驗。對離群值進行檢驗之前,需對檢測數據進行正態分布檢驗[14,22]。本研究為保證各實驗室的檢測值無異常值,依次采用了夏皮洛-威爾克檢驗、柯克倫檢驗及格拉布斯檢驗進行檢驗。 3.1.1 正態分布檢驗 檢驗偏離正態分布的方法有圖文法、有方向檢驗、多方向檢驗、夏皮洛-威爾克檢驗及愛潑斯—普利檢驗等,參考《數據的統計處理和解釋-正態性檢驗》[16],根據8家實驗室的檢測數據的情況,選取夏皮洛-威爾克檢驗,n=24且P=α=0.05時P的分位數為0.916,計算得W值為0.970>0.916,所以在顯著性水平α=0.05上不拒絕零假設,即數據符合正態分布。 3.1.2 柯克倫檢驗 參考《測量方法與結果的準確度第2部分:確定標準測量方法重復性與再現性的基本方法》[17],對8個參加協同定值的實驗室提供的數據進行柯克倫檢檢。柯克倫準則嚴格應用在所有標準差都是在重復性條件下,且由相同數目的測試結果計算的情形。給定P個由相同的n次重復測試結果計算的標準偏差Si(表6),在P=8,n=3時,柯克倫的臨界值為C0(1%)=0.615,C0(5%)=0.516,8個實驗室的柯克倫檢驗的結果為0.3060,即C=0.3060<0.516[C0(5%)],則8個實驗室檢測結果無異常值,均可參與后續檢驗。 3.1.3 格拉布斯檢驗 柯克倫所檢驗是對實驗室內變異的檢驗,僅對應于一組標準差中的最大值,即單側離群值檢驗,而格拉布斯檢驗主要是對實驗室間變異的檢驗,因此有必要依次對8個實驗室的均值分別進行單值和雙值格拉布斯檢驗[17]。 在P=8時,單值格拉布斯檢驗的臨界值G1(1%,8)=2.274,G1(5%,8)=2.126,而8家實驗室的單值格拉布斯檢驗結果G11=1.3206,G1p=1.5982,均 在P=8時,雙值格拉布斯檢驗的臨界值G2(1%,8)=0.0563,G2(5%,8)=0.1101,而雙值格拉布斯檢驗結果G21=0.4238,G2p=0.4127,均>G2(5%,8),因此所有單元的數據均為正確值,均可參與后續定值。 3.1.4 定值 數據經正態檢驗、柯克倫檢驗及格拉布斯檢驗,所有單元的數據均可保留并參與最終定值,定值結果為2.82 μg/kg。 特性值不確定度UCRM的評定參照《標準樣品工作導則(3)標準樣品定值的一般原則和統計方法》[18],由于本批次采用郵政網絡協同定值,因短期運輸所引起的不確定度usts已包含在各實驗室的檢測值中,不用再單獨計算。故本批次標準品的不確定度來源由3部分組成:測定值標準不確定度uchar、瓶間標準不確定度ubb、長期穩定性標準不確定度ults。 3.2.1 測定值標準不確定度uchar 包括實驗室檢測環境、儀器條件、檢測方法、檢測人員不同等產生的不確定度,各實驗室檢測結果平均值的標準偏差s=0.3392;即定值不確定度 3.2.2 瓶間標準不確定度ubb 瓶間不均勻性產生的不確定度 式中瓶間標準偏差的平方 重復性標準偏差 3.2.3 長期穩定性標準不確定度ults 本批次標準品進行了為期7個月的長期穩定性檢測(t=7),采用直線為經驗模型,假設特性值Y以初始值Y0以一個常數b1線性降解(b1為相對降解速率),為時間X的函數:Y(b0,b1,X)=Y0(1+b1X)。特性值的不確定度可通過將自變量Y0,X和b1的不確定度傳播給因變量Y得到[24,25]。 在沒有顯著降解的情況下,長期穩定性標準不確定度ults=s(b1)×t=0.136 μg/kg 從-18 ℃保存的樣本看未見微生物腐敗、變質情況出現,可保持7個月目標物長期穩定;協同定值是在常溫條件下將所制得的鰻鱺樣本通過郵政網絡傳遞給8家實驗室,從反饋的樣品接收狀態看,所有實驗室均未有諸如脹包、樣品變質等不正常樣品狀態的報告;樣品傳遞后的檢測結果也驗證了樣品中目標物的短期穩定性。綜合評估認為所制樣品可以常溫傳遞。故該標準物質的保存條件為-18 ℃冷凍密封保存,短期運輸條件(≤14 d)為常溫運輸,運輸時間較長時可以采用冰袋冷凍方式。 本研究采用液相色譜-串聯質譜法,多家實驗室郵政網絡協同定值,制得一批含有孔雀石綠代謝物隱性孔雀石綠染料殘留的鰻鱺肌肉標準物質。結果量值可靠,均勻性和穩定性良好,孔雀石綠代謝物隱性孔雀石綠特性值為(2.82±0.39) μg/kg(k=2)。本研究建立的研制和定值方法對進一步開展藥用染料殘留檢測用基體標準物質研制具有重要的意義。 [1] 全浩, 韓永志. 標準物質及其應用技術[M]. 2版.北京:中國標準出版社, 2002. [2] Zakaria O, Rezali M F. Reference materials as a crucial tools for validation and verification of the analytical process[J]. Procedia Soc Behavl Sci, 2014 ,121(2):204-213. [3] 徐向榮, 郝青, 彭加喜, 等. 水產品中殘留孔雀石綠研究進展[J]. 熱帶海洋學報, 2013,32 (4):97-106. [4] 張瀾瀾, 李桂偉, 花麗茹, 等. 禁用獸藥—孔雀石綠[J]. 黑龍江水產, 2015(1):43-44. [5] 張藝蓓, 岳田利, 喬海鷗, 等. 超高效液相色譜—串聯質譜法檢測魚中孔雀石綠、結晶紫及其代謝物[J].食品科學, 2014, 35(10):179-184. [6] Doerge D R, Chang H C, Divi R L, et al. Mechanism for inhibition of thyroid peroxidase by leucomalachite green [J]. Chem Res Toxicol, 1998, 11(9):1098-1104. [7] He H, Yang S, Yu K, et al. Microwave induced catalytic degradation of crystal violet in nano-nickel dioxide suspensions[J]. J Hazard Mater, 2010, 173(1/3):393-400. [8] Culp S J, Mellick P W, Trotter R W, et al. Carcinogenicity of malachite green chloride and leucomalachite green in B6C3F1 mice and F344 rats[J]. Food Chem Toxicol, 2006, 44(8):1204-1212. [9] Srivastava S, Sinha R, Roy D. Toxicological effects of malachite green[J]. Aquat Toxicol, 2004, 66(3):319-329. [10] 朱萬燕, 劉玉芳, 劉冰, 等. 同位素內標法測定水產品中孔雀石綠、結晶紫及其代謝產物[J]. 水產科學, 2011, 30(12):781-784. [11] 楊方, 楊守深, 盧聲宇, 等. 鰻鱺肌肉凍干粉中呋喃唑酮代謝物3-氨基-2-(噁)唑烷酮殘留標準物質的研制[J]. 分析化學, 2010, 38(3):397-400. [12] 余孔捷, 楊方, 劉正才, 等. 含氟甲喹殘留的鰻鱺肉糜自然基體標準樣品的制備[J]. 色譜, 2011, 29(7):691-695. [13] 時文春, 馮燕平, 李開鋒, 等. 魚肉中氯霉素標準樣品的研制[J]. 分析測試學報, 2012, 31(10):1339-1344. [14] 黃超群, 謝文, 侯建波, 等. 蜂王漿凍干粉中甲硝唑殘留標準樣品制備的研究[J]. 分析測試學報, 2013, 32(4):512-518. [15] 余孔捷, 劉正才, 黃杰, 等. 含喹諾酮類多殘留鰻糜自然基體標準樣品快速制備[J]. 分析試驗室, 2015, 34(2):182-185. [16] 國家質量技術監督局. GB/T 4882—2001,數據的統計處理和解釋—正態性檢驗[S].北京:中國標準出版社,2001. [17] 中國標準化研究院. GB/T6379.2—2004,測量方法與結果的準確度第2部分:確定標準測量方法重復性與再現性的基本方法[S]. 北京:中國標準出版社,2004. [18] 全國標準樣品技術委員會. GB/T 15000.3—2008,標準樣品工作導則(3)標準樣品定值的一般原則和統計方法[S].北京:中國標準出版社,2008. [19] 韋英亮, 潘艷坤. 標準物質期間核查溯源性定值分析方法[J]. 化學分析計量, 2012, 21(5):101-103. [20] 盧曉華. 標準物質的溯源性與分級[J]. 中國計量, 2007(7):39-41. [21] 全國水產標準化技術委員會. GB/T 19857—2005,水產品中孔雀石綠和結晶紫殘留量的測定[S].北京:中國標準出版社,2005. [22] 韓永志, 韓冰. 標準物質量值的溯源性及不確定度[J]. 化工標準·計量·質量, 2002(10):22-24. [23] 楊麗君, 梁君妮, 曹鵬, 等. 魚肉中氟苯尼考標準樣品的制備[J]. 食品科學, 2015, 36(14):176-180. [24] Van der Veen A M H, Linsinger T P J, Schimmel H, et al. Uncertainty calculations in the certification of reference materials 4. Characterisation and certification[J]. J Accred Qual Assur, 2001, 6(7):290-294. [25] Linsinger T P J, Pauwels J, Lamberty A, et al. Estimating the uncertainty of stability for matrix CRMs[J]. Fresenius J Anal Chem,2001, 370(2/3):183-188. DevelopmentofaReferenceMaterialforLeucomalachiteGreenResiduesinEelMuscle YIN Taikun1,YANG Fang2,LIU Zhengcai2,LIN Yonghui2 ( 1. School of Chemical Engineering, Fuzhou University, Fuzhou 350108, China; 2. Technology Centre of Fujian Entry-exit Inspection and Quarantine Bureau, Fuzhou 350001, China ) Eel (Anguillaanguilla) was bathed in malachite green at certain concentration to make the malachite green enter the eel and contain leucomalachite green (LMG) in muscle via metabolism to establish a method for the preparation and certification of the reference material of leucomalachite green (LMG) residues in the muscle. The positive 500 bags of samples of eel were performed in one batch by the procedure of homogenation, vacuum packing and irradiation. The homogeneity and stability of samples were detected by ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS). The concentration of the chemical constituent of the sample was certified through the collaborative analysis program participated by 8 laboratories, and the uncertainty assessment was performed with property value of 2.82 μg/kg, and the expanded uncertainty of 0.39 μg/kg(k=2). It is concluded that the preparation method for matrix reference materials for the dye residues provides one kind of reference. eel muscle; leuco malachite green; residue ; reference material 10.16378/j.cnki.1003-1111.2016.03.014 S912 A 1003-1111(2016)03-0272-06 2015-08-24; 2015-10-08. 質檢公益項目(201310143-02);福建省科技重點項目(2012Y6001). 尹太坤(1988-),男,碩士研究生;研究方向:食品安全分析.E-mail:1257005106@qq.com.通訊作者: 楊方(1969-),女,主任技師;研究方向:農獸藥殘留檢測.E-mail:yangf@fjciq.gov.cn.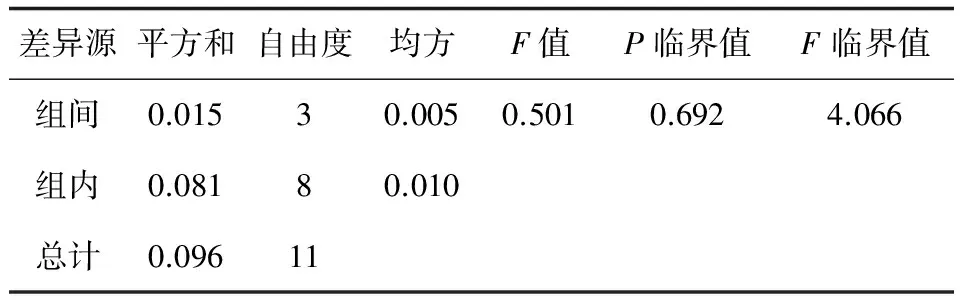
3 討 論
3.1 協同定值與離群值檢驗
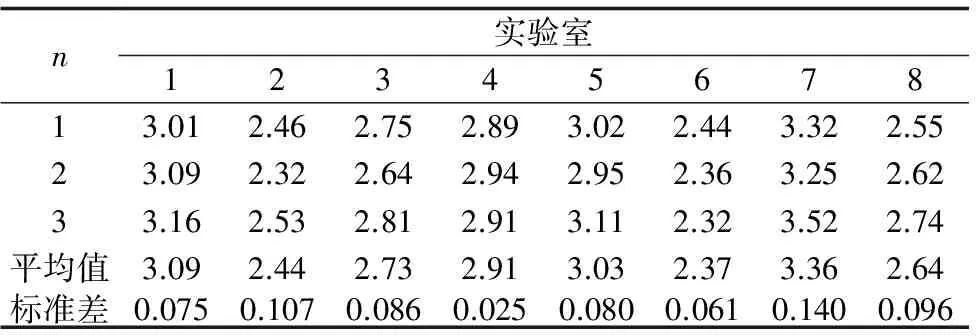
3.2 不確定度評定
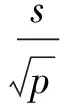
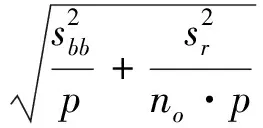
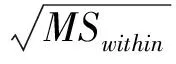
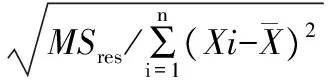
3.3 日常使用運輸與儲存條件
4 結 論
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58當代陜西(2019年8期)2019-05-09 02:22:48動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10電子競技(2019年22期)2019-03-07 05:17:26電子競技(2019年21期)2019-02-24 06:55:52電子競技(2019年20期)2019-02-24 06:55:35電子競技(2019年19期)2019-01-16 05:36:09
