近岸及河口海水中全氟化合物的固相萃取富集/超高效液相色譜-串聯質譜測定
2016-12-20 02:57:42黃東仁溫裕云陳志華李榮麗弓振斌
分析測試學報 2016年3期
黃東仁,溫裕云,陳志華,李榮麗,弓振斌*
(1.福建省海洋環境與漁業資源監測中心,福建 福州 350003;2.福建遠東技術服務有限公司,福建 泉州 362006;3.廈門大學 環境與生態學院,福建 廈門 361102)
?
近岸及河口海水中全氟化合物的固相萃取富集/超高效液相色譜-串聯質譜測定
黃東仁1,溫裕云2,陳志華2,李榮麗3,弓振斌3*
(1.福建省海洋環境與漁業資源監測中心,福建 福州 350003;2.福建遠東技術服務有限公司,福建 泉州 362006;3.廈門大學 環境與生態學院,福建 廈門 361102)
建立了近岸及河口海水中全氟辛基磺酸(PFOS)、全氟辛酸(PFOA)、全氟十一酸(PFUnA)、全氟十二酸(PFDoA)、全氟十三酸(PFTrDA)、全氟十四酸(PFTA) 6種全氟化合物(PFCs)的超高效液相色譜-串聯質譜(UHPLC-MS/MS)測定方法。使用C18固相萃取小柱對500 mL水樣中的目標物進行富集后,用15 mL甲醇-乙酸乙酯混合淋洗液(4∶1)進行洗脫,濃縮,定容至1.0 mL后,用Kinetex XB-C18色譜柱以均含5.0 mmol/L甲酸銨的甲醇-水為流動相梯度洗脫方式進行分離,電噴霧負離子模式(ESI-)電離,多重反應監測模式(MRM)以及內標法對6種PFCs進行定性定量測定。優化了固相萃取、色譜分離及質譜測定條件,考察了海水鹽度對方法回收率的影響。在優化實驗條件下,方法在2.0,5.0,10.0 ng/L加標水平下,實際海水樣品的回收率為80.1%~117.4%,在2.0 ng/L加標水平的相對標準偏差(RSD,n=7)為8.2%~12.1%。6種PFCs的線性范圍為0.5~50.0 μg/L,相關系數大于0.999 0;方法的定量下限(LOQ,S/N=10)為0.5~1.5 ng/L。該方法具有樣品前處理簡單、分析速度快、選擇性好的特點,適用于近岸及河口海水中全氟化合物的快速測定。
全氟化合物;近岸及河口海水;液相色譜-串聯質譜;固相萃取
全氟化合物(Perfluorinated compounds,PFCs)是直鏈或支鏈烷烴中的氫原子全部被氟取代后的化合物,因具有優良的化學穩定性、熱穩定性、高表面活性及疏水疏油性,被廣泛用于化工、紡織、皮革、消防、洗滌劑、食品包裝、裝潢等行業[1-3]。但PFCs具有環境穩定、污染范圍廣、高生物累積等持久性有機物(POPs)的特性,已引起國際社會的高度關注[2],許多國家和地區頒布法律/法規限制PFCs在各種產品中的使用。全氟辛基磺酸(PFOS)于2009 年9 月列入POPs 公約附表B,在全球范圍內限制生產與使用[2]。同時歐盟也對進口紡織品和皮革產品中的PFOS 殘留量進行限制[4-5]。美國環境保護署(U.S.EPA)要求至2010年削減95%全氟辛酸(PFOA)及長鏈PFCs的生產量和產品殘留量,至2015年完全禁止生產與使用PFOA 及長鏈PFCs[6]。
水體是PFCs在環境中遷移、轉化的重要載體,也是污染受體和歸宿。近岸、河口位于海-陸交匯帶,人類活動的主要陸源污染物如PFCs經近岸排污、河口徑流輸入后進入海洋水體;據報道[7-8],近岸、河口水體中PFCs的含量大多在幾至幾十ng/L之間,中位濃度在2~10 ng/L之間,其中PFOS和PFOA是最常檢出的組分,且其含量遠高于其它組分[9-14]。因此,建立近岸、河口水體中PFCs的準確、快速測定方法,對了解PFCs在環境中的遷移轉化、污染控制以及環境保護等具有重要意義。
水體中PFCs的測定方法主要有氣相色譜-質譜法(GC-MS)[15-16]、液相色譜-質譜法(LC-MS)[17-20]。但由于PFCs具有高沸點、難揮發的特點,使用GC-MS測定時需通過衍生使PFCs酯化;而LC-MS尤其是使用串聯質譜(MSn)檢測器時,無需衍生化,且檢測靈敏度高,定性定量能力強,已被廣泛應用于各種新型污染物的監測。目前已報道的研究工作主要集中于地表水中PFOS和PFOA的測定,對中鏈和長鏈PFCs的同時測定方法僅有少量報道[20],而針對近岸及河口高鹽度海水或淡咸水中多種PFCs的測定方法國內鮮有報道[18]。本研究通過對色譜-串聯質譜條件、固相萃取前處理等過程進行優化,建立了C18固相萃取(SPE)富集、超高效液相色譜-串聯質譜(LC-MS/MS)快速測定近岸及河口海水中PFOS、PFOA、全氟十一酸(PFUnA)、全氟十二酸(PFDoA)、全氟十三酸(PFTrDA)、全氟十四酸(PFTA)的分析方法。
1 實驗部分
1.1 儀器與試劑
Agilent 1290液相色譜儀(美國安捷倫科技有限公司);Agilent 6460三重串聯四極桿質譜儀(美國安捷倫科技有限公司),系統配置Agilent JetStream Technologies(AJS) 電噴霧離子源(ESI)接口、MassHunter工作站軟件。Milli-Q超純水制備系統(美國Millipore公司);BHA-9050A濃縮儀(Caliper Life Sciences,USA);0.22 μm一次性針式過濾膜(天津市津騰實驗設備有限公司);C18固相萃取柱(500 mg/6 mL,BESEP,北京振翔科技有限公司)。
甲醇、乙腈(美國Merck公司),甲酸、甲酸銨(美國Sigma公司),純度均為LC-MS級;6種全氟化合物標準品(純度>98.0%,Dr.Ehrenstorfer GmbH);內標化合物13C4-全氟辛基磺酸(MPFOS,1.2 mL,50 mg/L)、13C4-全氟辛酸(MPFOA,1.2 mL,50 mg/L)購于Wellington Laboratories公司。
稱取100 mg單一標準品(固體)于10 mL容量瓶,用甲醇溶解并定容,配成濃度為10.0 mg/mL的單標儲備液;液體標準品則用甲醇稀釋成合適濃度的儲備液,使用時再以甲醇-水流動相(60∶40)逐步稀釋至所需濃度。
1.2 樣品采集與前處理
海水樣品采取后在現場過濾。濾膜為0.45 μm玻璃纖維,過濾后使水樣沿瓶壁緩慢注入1 L 棕色磨口玻璃瓶,并用鋁箔和棉線扎緊瓶塞密封,于冷藏箱中保存、運輸至實驗室并盡快分析,或保存于4 ℃冰箱中,最長保存24 h。
C18固相萃取小柱使用前,依次用6 mL正己烷、丙酮、甲醇、Milli-Q純水進行活化,待用。
量取500 mL過濾后的水樣并加入0.5 mL的5.0 μg/L內標混合溶液,用活化后的C18固相萃取柱進行基體分離及目標物富集,流速5 mL/min,水樣全部流過后再真空抽干固相萃取柱約45 min,然后用15 mL甲醇-乙酸乙酯混合淋洗液(4∶1)洗脫,收集流出液并濃縮至近干,以甲醇定容至1.0 mL,待上機測試。
為避免器皿引入較高空白,實驗過程避免使用PTFE材質的器皿和色譜管路,本工作使用玻璃器皿以及PEEK或不銹鋼管路。為保證實驗數據質量,測定每批樣品時必須做方法流程空白。
1.3 色譜分離及質譜測定條件
色譜柱Phenomenex Kinetex XB-C18(2.1 mm×50 mm,2.6 μm);流動相:A為甲醇(含5.0 mmol/L甲酸銨),B為水(含5.0 mmol/L甲酸銨),流速0.4 mL/min,柱溫40 ℃;梯度洗脫程序為:0~0.5 min,60%~80% A,0.5~1.9 min,80%~90% A,1.9~2.5 min,90% A;進樣量2.0 μL。
電噴霧采用負離子模式(ESI-);干燥氣溫度350 ℃,流速8 L/min;霧化氣壓力:241.3 kPa;鞘氣溫度380 ℃,流速8 L/min;毛細管電壓:4 kV;噴嘴電壓:3.5 kV。定性/定量采用多重反應監測(MRM)模式,參數見表1。內標法定量。
表1 待測目標物及內標化合物的MRM分析參數
Table 1 MRM conditions for target analytes and internal standards
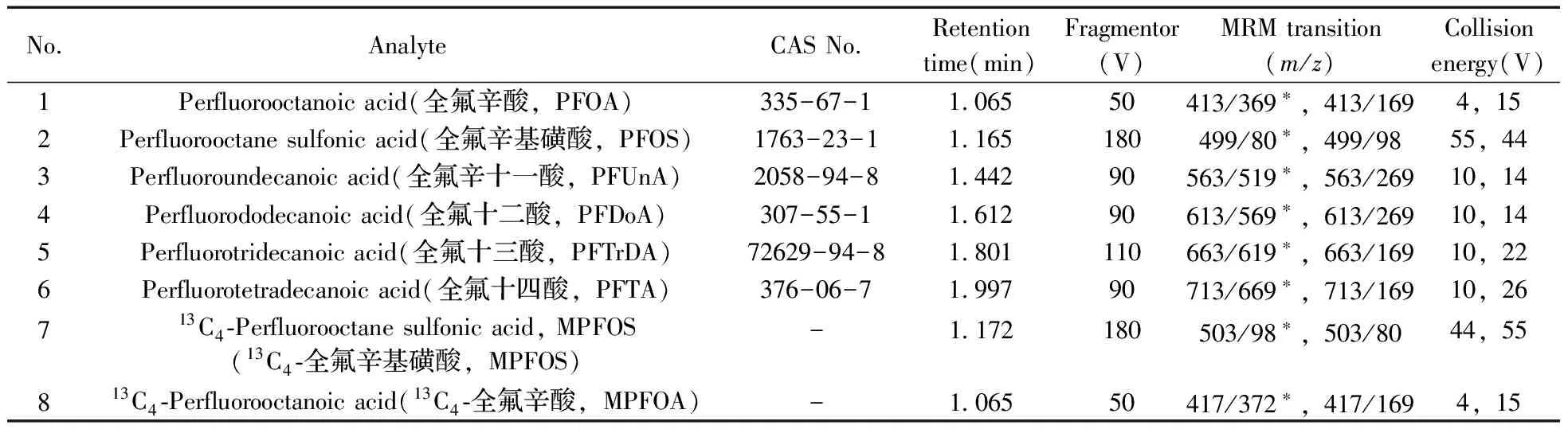
No.AnalyteCASNo.Retentiontime(min)Fragmentor(V)MRMtransition(m/z)Collisionenergy(V)1Perfluorooctanoicacid(全氟辛酸,PFOA)335-67-1106550413/369?,413/1694,152Perfluorooctanesulfonicacid(全氟辛基磺酸,PFOS)1763-23-11165180499/80?,499/9855,443Perfluoroundecanoicacid(全氟辛十一酸,PFUnA)2058-94-8144290563/519?,563/26910,144Perfluorododecanoicacid(全氟十二酸,PFDoA)307-55-1161290613/569?,613/26910,145Perfluorotridecanoicacid(全氟十三酸,PFTrDA)72629-94-81801110663/619?,663/16910,226Perfluorotetradecanoicacid(全氟十四酸,PFTA)376-06-7199790713/669?,713/16910,26713C4?Perfluorooctanesulfonicacid,MPFOS(13C4?全氟辛基磺酸,MPFOS)-1172180503/98?,503/8044,55813C4?Perfluorooctanoicacid(13C4?全氟辛酸,MPFOA)-106550417/372?,417/1694,15
*quantitative ion pair
2 結果與討論
2.1 樣品前處理條件的優化
水體中PFCs的分離富集方法主要采用固相萃取,萃取柱填料為石墨化碳黑、C18、HLB、WAX等[17-19,21]。本工作分別選用石墨化碳黑、C18、HLB 3種常見的萃取柱進行實驗。結果表明,石墨化碳黑對6種PFCs的萃取效果較好,但對于長鏈組分如PFTrDA和PFTA則難以使用甲醇、乙腈等常用試劑進行洗脫,且回收率偏低;HLB柱也能較好地萃取待測PFCs,但采用甲醇或乙腈洗脫時某些PFCs的回收率偏低;C18柱對待測6種PFCs的萃取效果好,通過調整洗脫液(甲醇-乙酸乙酯)的比例,所有目標物均能獲得較高的回收率。因此,最終選用C18萃取柱,以甲醇-乙酸乙酯(4∶1)混合溶液為洗脫液進行實驗。
2.2 色譜-質譜條件的選擇
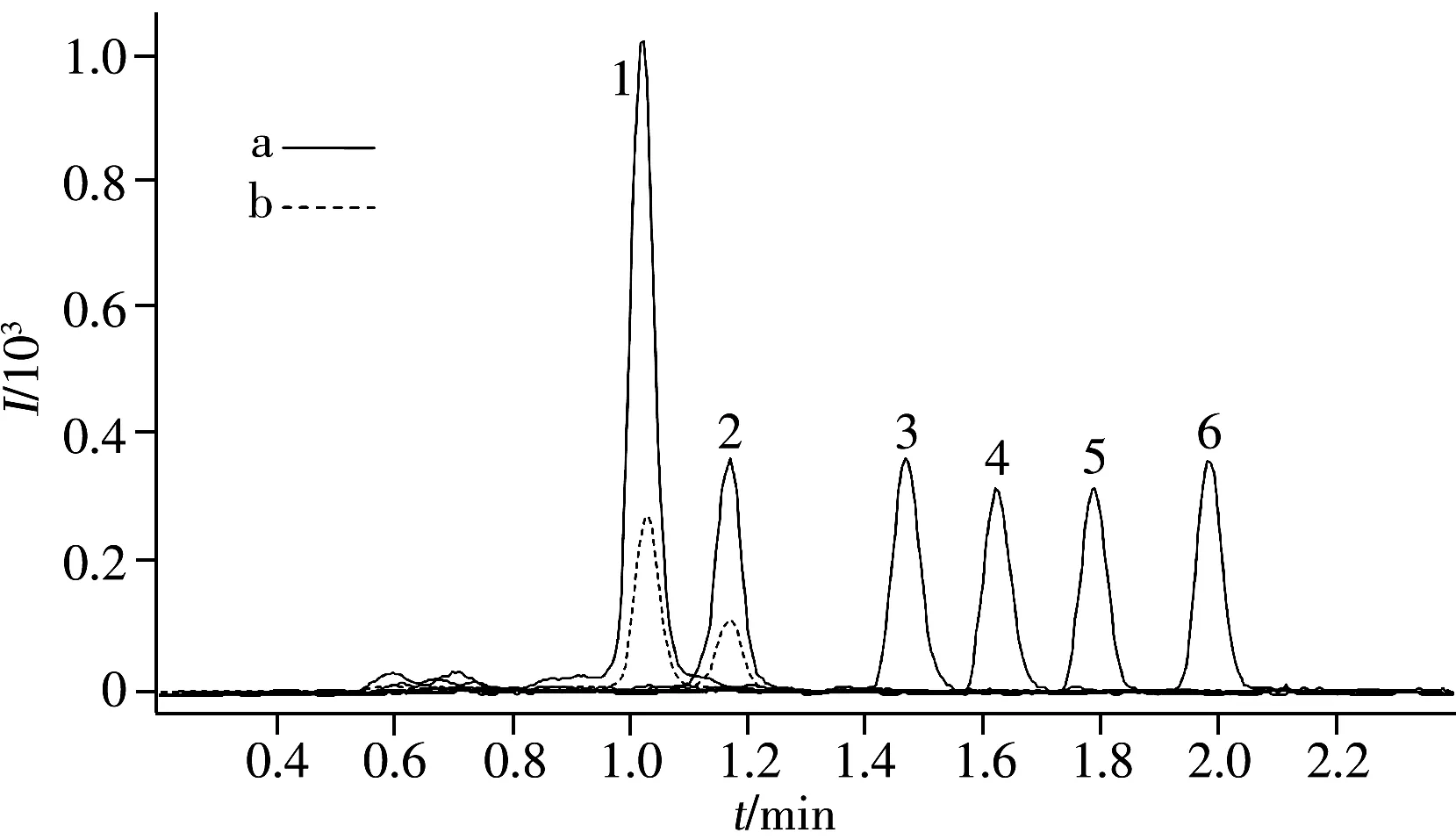
圖1 空白海水樣品加標濃度2.0 ng/L(a) 與 6#海水樣品(b)的 MRM色譜圖Fig.1 MRM Chromatograms of blank seawater sample spiked with 2.0 ng/L(a) and real seawater sample of 6#(b) 1.PFOA,2.PFOS,3.PFUnA,4.PFDoA,5.PFTrDA,6.PFTA
實驗選擇對中等極性、弱極性待測組分有良好保留特性及分離能力的Kinetex XB-C18色譜柱,分別采用甲醇-水(兩相中均含0.01%,0.05%,0.1%甲酸以及兩相中均含1.0,2.5,5.0,10.0 mmol/L甲酸銨)、乙腈-水(2.0 mmol/L甲酸銨)流動相進行梯度洗脫,考察不同流動相種類的影響。結果表明,上述體系均能使目標組分得到良好分離,但甲醇-水體系中加入甲酸后待測組分的質譜響應有所下降;而含有甲酸銨的甲醇-水流動相體系不但使PFCs的質譜響應明顯增加,而且可改善色譜峰形。綜合考慮,實驗選擇甲醇-水(甲醇和水中均含5.0 mmol/L甲酸銨)體系進行分離,6種目標組分可在2.1 min內被洗脫。色譜分離結果見圖1a。
采用儀器自帶的優化軟件Optimizer對碎裂電壓(Fragmentor)、碰撞能量(Collision energy,CE)、定性/定量MRM離子對進行優化,使用直接進樣方式,以甲醇-水(60∶40,含5.0 mmol/L甲酸銨)為流動相,得到各待測組分的最佳MRM采集參數,如表1所示。實驗還優化了干燥氣溫度、干燥氣流速、霧化氣壓力、鞘氣溫度、鞘氣流速、毛細管電壓、噴嘴電壓等離子源參數,使大多數組分得到最佳的信號響應。
2.3 鹽度對回收率的影響
在不同的離子強度下,目標化合物在水相和有機相中的分配行為可能會發生變化,從而影響SPE柱的萃取效率和洗脫液的洗脫過程。實驗考察了海水鹽度對加標回收率的影響,將鹽度為35的海水樣品稀釋為鹽度(‰)分別為0,7,14,21,28,35的海水,然后加入混合標準溶液,使各待測組分的濃度均為2.0 ng/L,依照本方法進行測定,計算不同鹽度樣品中各組分的回收率(如表2)。結果表明,不同鹽度下各目標組分回收率的相對標準偏差(RSD)為1.7% ~ 7.1%,表明鹽度對回收率無顯著影響。但在測定碳鏈長大于12以上的PFCs時,回收率明顯下降,為提高回收率,可采用相應目標物的同位素標記物(如13C標記)作為替代物進行實驗。因此,本方法適用于近岸特別是河口區鹽度變化較大的海水中PFCs的準確測定。
表2 不同鹽度下樣品的加標回收率
Table 2 Effect of seawater salinity on recoveriesof PFCs spiked at 2.0 ng/L level
(%)
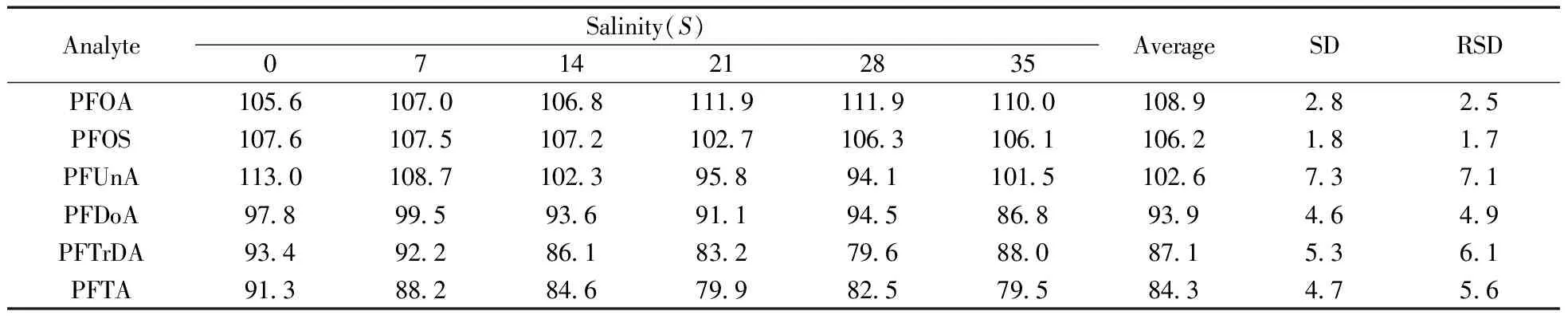
AnalyteSalinity(S)0714212835AverageSDRSDPFOA10561070106811191119110010892825PFOS10761075107210271063106110621817PFUnA113010871023958941101510267371PFDoA9789959369119458689394649PFTrDA9349228618327968808715361PFTA9138828467998257958434756
2.4 精密度與加標回收率
以實驗室制備的模擬樣品進行方法的精密度實驗。在實際海水樣品中分別添加濃度水平為2.0 ng/L的混合標準溶液,放置24 h后按本方法平行測定7次,結果見表3。其相對標準偏差(RSD)為8.2%~12.1%,方法的精密度可滿足近岸、河口海水中6種PFCs的測定要求。
在空白海水樣品中分別添加濃度水平為2.0,5.0,10.0 ng/L的混合標準溶液及內標溶液,放置24 h后依照本方法進行測定,結果如表3所示。在3個加標水平下,實際樣品的回收率為80.1%~117.4%,方法的準確度可滿足實際海水樣品測定的需要。
表3 PFCs在不同濃度加標水平下的回收率及相對標準偏差
Table 3 Recoveries and RSDs of real samples spiked with different levels
(%)
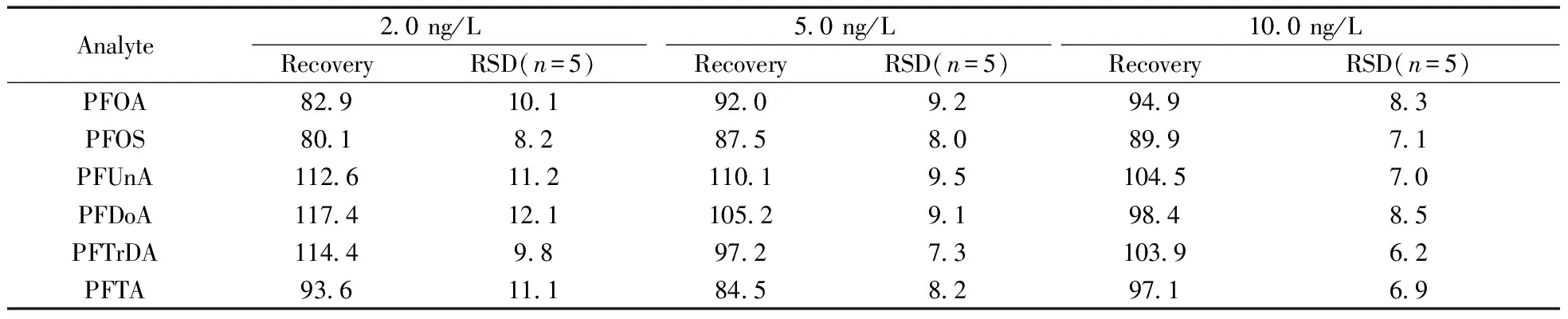
Analyte20ng/L50ng/L100ng/LRecoveryRSD(n=5)RecoveryRSD(n=5)RecoveryRSD(n=5)PFOA8291019209294983PFOS801828758089971PFUnA1126112110195104570PFDoA117412110529198485PFTrDA11449897273103962PFTA9361118458297169
2.5 線性范圍與定量下限
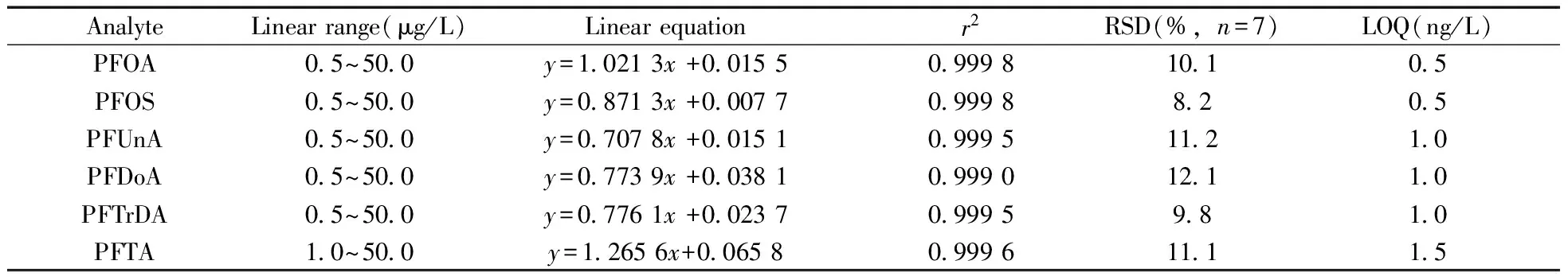
配制濃度為0.5,2.0,5.0,10.0,20.0,50.0 μg/L 的PFCs系列混合標準溶液,其內標濃度為2.5 μg/L(PFOS以MPFOS為內標,其它組分以MPFOA為內標),在優化條件下進行測定,以待測組分濃度(x,μg/L)為橫坐標,定量離子對的峰面積(yi)與相應內標物峰面積(yin)的比值(y)為縱坐標進行線性回歸,結果見表4。結果表明,待測的6種PFCs在上述濃度范圍內有良好的線性關系,相關系數均大于0.999 0。
表4 方法的線性關系、相對標準偏差及定量下限
Table 4 Linearity,RSDs and limits of quantitation(LOQs)
AnalyteLinearrange(μg/L)Linearequationr2RSD(%,n=7)LOQ(ng/L)PFOA05~500y=10213x+001550999810105PFOS05~500y=08713x+00077099988205PFUnA05~500y=07078x+001510999511210PFDoA05~500y=07739x+003810999012110PFTrDA05~500y=07761x+00237099959810PFTA10~500y=12656x+006580999611115
在空白水樣中從高到低添加待測組分后進行檢測,直到獲得信噪比等于10(S/N=10)的濃度,確定其為方法的定量下限(LOQ),其中噪聲的測量時間為0.1 min,噪聲測量方式為均方根(RMS),由儀器自帶工作站軟件計算所得[22]。表4結果表明,6種PFCs的LOQ為0.5~1.5 ng/L。
2.6 實際樣品的測定
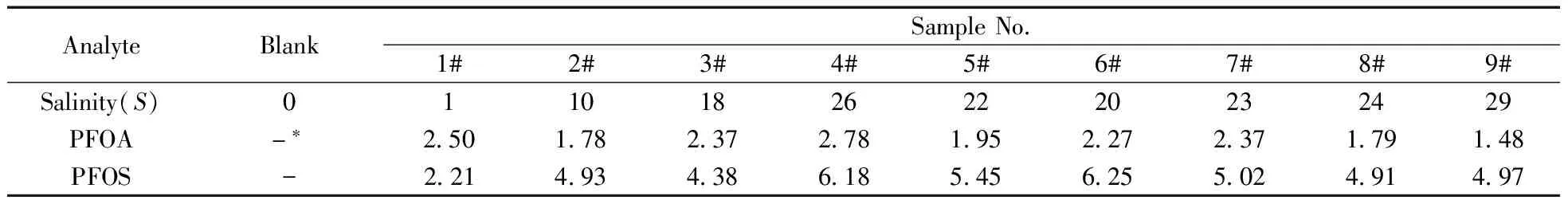
采用本方法,對2014年9月采自廈門九龍江河口區的表層海水樣品進行分析,其鹽度及測定結果如表5所示,圖1b為6#海水樣品的色譜圖。結果表明,所有9個河口表層海水樣品中均檢出PFOA和PFOS;除1#樣品外,其它樣品中PFOS的濃度均明顯高于PFOA;另外4種組分在采集的海水樣品中均未檢出,表明其濃度低于方法的定量下限。
表5 廈門九龍江口實際樣品的測定結果
Table 5 Analytical results of samples of Jiulongjiang River in Xiamen
ρ/(ng·L-1)
*:no detected
3 結 論
本文通過對固相萃取分離富集過程、色譜-質譜條件等的優化,建立了超高效液相色譜-串聯質譜快速測定近岸及河口水體中全氟化合物的方法,并應用于實際樣品的檢測。方法具有簡單、快速、準確等特點,可為近岸、河口區海水中全氟化合物的海洋環境化學研究提供技術基礎。
[1] Zareitalabad P,Siemens J,Hamer M,Amelung W.Chemosphere,2013,91(6):725-732.
[2] Wang Y W,Cai Y Q,Jiang G B.Sci.Sin.Chim.(王亞韡,蔡亞岐,江桂斌.中國科學:化學),2010,40(2):90-123.
[3] Li J,Zhang H,Chai Z F,Shen J C,Yang B.J.Instrum.Anal.(李靜,張鴻,柴之芳,沈金燦,楊波.分析測試學報),2014,(10):1109-1115.
[4] Huang X L,Wu H Q,Huang F ,Lin X S ,Zhu Z X.Chin.J.Anal.Chem.(黃曉蘭,吳惠勤,黃芳,林曉珊,朱志鑫.分析化學),2007,35(11):1591-1595.
[5] Zhang X L,Liu C H,Xian Y P,Mai X X.J.Instrum.Anal.(張曉利,劉崇華,冼燕萍,麥曉霞.分析測試學報),2010,29(12):1182-1185.
[6] Hogue C.Chem.Eng.News,2006,84(10):10.
[7] Ahrens L.J.Environ.Monit.,2011,13(1):20-31.
[8] So M K,Taniyasu S,Yamashita N,Giesy J P,Zheng J,Fang Z,Im S H,Lam P K S.Environ.Sci.Technol.,2004,38(15):4056-4063.
[9] Liu B L,Zhang H,Xie L W,Liu G Q,Wang Y P,Wang X X,Li J,Dong W H.Environ.Sci.(劉寶林,張鴻,謝劉偉,劉國卿,王艷萍,王鑫璇,李靜,董煒華.環境科學),2015,(6):2028-2037.
[10] Chen Q W,Zhang H,Chai Z F,Shen J C,Yang B.Environ.Sci.(陳清武,張鴻,柴之芳,沈金燦,楊波.環境科學),2012,33(6):1795-1800.
[11] Zhao S J,Cao P,Zhu C K,Yan Y Q,Shen J F,Yan H T,Yang X J.Mar.Environ.Sci.(趙淑江,曹培,朱誠侃,顏雅琴,沈娟芳,閆洪濤,楊新軍.海洋環境科學),2012,(2):221-224.
[12] Cai M H,Zhao Z,Yang H Z,Yin Z G,Hong Q Q,Sturm R,Ebinghaus R,Ahrens L,Cai M G,He J F,Xie Z Y.Environ.Pollut.,2012,161:162-169.
[13] Wang T Y,Lu Y L,Chen C L,Naile J E,Khim J S,Park J,Luo W,Jiao W T,Hu W Y,Giesy J P.Mar.Pollut.Bull.,2011,62(8):1905-1914.
[14] Chen C L,Wang T,Khim J S,Luo W,Jiao W T,Lu Y L,Naile J E,Hu W Y,Zhang X,Geng J,Bi C C,Li J,Giesy J P.Chem.Ecol.,2011,27:165-176.
[15] Martin J W,Muir D C G,Moody C A,Ellis D A,Kwan W C,Solomon K R,Mabury S A.Anal.Chem.,2002,74(3):584-590.
[16] Langlois I,Berger U,Zencak Z,Oehme M.RapidCommun.MassSpectrom.,2007,21(22):3547-3553.
[17] Zhang P,Shi Y L,Cai Y Q,Mou S F.Chin.J.Anal.Chem.(張萍,史亞利,蔡亞岐,牟世芬.分析化學),2007,35(7):969-972.
[18] Li X,Chen J H,Cheng H Y,Shi Q,Jiang F H,Zheng L,Wang X R.Environ.Chem.(李鑫,陳軍輝,程紅艷,史倩,蔣鳳華,鄭立,王小如.環境化學),2012,31(6):896-901.
[19] Liu L Z,Zeng T,Peng R F,Lin Y N,Luo X Y.Chin.J.HealthLab.Technol.(劉莉治,曾濤,彭榮飛,林玉娜,羅曉燕.中國衛生檢驗雜志),2013,23(8):1831-1834.
[20] Liu Q,He D C,Xu Z C,Li J,Zhang S K,Pan L.Environ.Monit.China(劉慶,賀德春,許振成,李杰,張素坤,潘浪.中國環境監測),2014,30(4):134-139.
[21] Wang C,Lü Y B,Chen H J,Tan L,Teng E J.Chin.J.Chromatogr.(王超,呂怡兵,陳海君,譚麗,滕恩江.色譜),2014,32(9):919-925.
[22] Agilent Technology.UnderstandingYourChemStation.Germany 07/2008,G2070-91125:232-253.
Rapid Analysis of Perfluorinated Compounds in Coastal and Estuarine Seawater by SPE Enrichment with Ultra High Performance Liquid Chromatography-Tandem Mass Spectrometry
HUANG Dong-ren1,WEN Yu-yun2,CHEN Zhi-hua2,LI Rong-li3,GONG Zhen-bin3*
(1.Monitoring Center of Marine Environment and Fishery Resources,Fuzhou 350003,China;2.PFI Fareast Testing & Technology Services Co.Ltd.,Quanzhou 362006,China;3.College of the Environment & Ecology,Xiamen University,Xiamen 361102,China)
A rapid method was developed for the quantitative analysis of perfluorinated compounds(PFCs) including perfluorooctane sulfonic acid(PFOS),perfluorooctanoic acid(PFOA),perfluoroundecanoic acid(PFUnA),perfluorododecanoic acid(PFDoA),perfluorotridecanoic acid(PFTrDA),perfluorotetradecanoic acid(PFTA) in coastal and estuarine seawater by solid phase extraction(SPE) enrichment with ultra high performance liquid chromatography tandem electrospray ionization mass spectrometry(UHPLC-MS/MS).Seawater sample was enriched and purified through a 500 mg C18SPE cartridge at 5.0 mL/min,eluted with 15 mL mixed solvent of methanol and ethyl acetate(4∶1),and finally concentrated into 1.0 mL with an automated de-solvent device.Target analytes were separated on a Kinetex XB-C18column using methanol(containing 5.0 mmol/L NH4COOH-water(containing 5.0 mmol/L NH4COOH) as mobile phase.The negative electrospray ionization(ESI-) and multiple reaction monitoring mode(MRM) were utilized.An internal standard calibration method was chosen for the quantitative analysis.The calibration curves of PFCs were linear in the range of 0.5-50.0 μg/L with correlation coefficients more than 0.999 0.The limits of quantitation(LOQs,S/N=10) were between 0.5 ng/L and 1.5 ng/L.The recoveries of 6 PFCs at three spiked levels(2.0,5.0,10.0 ng/L) were in the range of 80.1%-117.4%,and relative standard deviations(RSDs) were 8.2%-12.1% at spiked level of 2.0 ng/L.
perfluorinated compounds; coastal and estuarine seawater; ultra high performance liquid chromatography tandem mass spectrometry(UHPLC-MS/MS); solid phase extraction
2015-07-19;
2015-09-18
福建省自然科學基金重點項目(2012Y0005);環保公益性行業科研專項(201309007)
研究簡報
10.3969/j.issn.1004-4957.2016.03.008
O657.63; TL281
A
1004-4957(2016)03-0305-06
*通訊作者:弓振斌,博士,教授,研究方向:分析化學與環境化學,Tel:0592-2186572,E-mail:zbgong@xmu.edu.cn
