慢性阻塞性肺疾病大鼠肺組織JNK/SAPK水平變化及意義
2017-01-06 03:12:33田雪張杏怡李鋒肖輝周新
山東醫藥 2016年45期
田雪,張杏怡,李鋒,肖輝,周新
(上海交通大學附屬第一人民醫院,上海200080)
?
慢性阻塞性肺疾病大鼠肺組織JNK/SAPK水平變化及意義
田雪,張杏怡,李鋒,肖輝,周新
(上海交通大學附屬第一人民醫院,上海200080)
目的 探討C-Jun氨基末端蛋白激酶(JNK)/應激激活蛋白激酶(SAPK)在慢性阻塞性肺疾病(COPD)大鼠肺組織中的水平變化及意義。方法 16只Ⅱ級Wistar雄性大鼠隨機分為對照組和COPD組。對照組不予任何處理,COPD組給予煙熏刺激,造成大鼠COPD動物模型。造模成功后,光鏡下觀察肺組織病理,動物肺功能檢測儀檢測肺功能,ELISA法檢測大鼠肺組織TNF-α水平,免疫組化法檢測肺組織JNK/SAPK表達,RT-PCR檢測肺組織JNK/SAPK mRNA表達水平。結果 COPD組光鏡下出現了大量炎性細胞浸潤,部分肺泡融合,肺泡數量減少,證明COPD造模成功。與對照組比較,COPD組第0.3秒用力呼氣容積與用力呼氣容積的比值降低,呼氣阻力、吸氣阻力升高,動態順應性下降(P<0.05或<0.01),TNF-α升高(P<0.05);免疫組化結果顯示,COPD組肺組織中JNK/SAPK蛋白表達高于對照組(P均<0.05)。RT-PCR結果顯示,與對照組相比,COPD組JNK/SAPK mRNA水平升高(P均<0.05)。JNK/SAPK mRNA表達水平與TNF-α呈正相關(r=0.751,P<0.05)。結論 煙霧誘導的COPD大鼠JNK/SAPK水平升高,其可能通過激活煙霧誘導的炎癥因子,導致氣道炎癥和肺氣腫,促進COPD發生。
慢性阻塞性肺疾病;C-Jun氨基末端蛋白激酶;應激激活蛋白激酶;作用機制
C-Jun氨基末端蛋白激酶(JNK)/應激激活蛋白激酶(SAPK)是一類絲氨酸/蘇氨酸激酶,為哺乳動物絲裂素活化蛋白激酶家族中的一員。胞外多種刺激信號,如生長因子、細胞因子、環境應激,促使JNK/SAPK磷酸化,使其從細胞質中移位到細胞核,與其下游核轉錄因子NF-κB、ATF2或c-Jun的氨基末端區域結合,使轉錄因子的活性區域發生磷酸化,促進相關基因表達,提高核轉錄能力,介導細胞生長、基因轉化、細胞分化和細胞凋亡等多種生物學效應[1]。JNK/SAPK被激活后,能通過不同的下游分子調控機體和細胞的各種功能,如mRNA轉錄、蛋白合成、線粒體功能等,其中很重要的一項功能是調控細胞自噬[2,3]。目前,在腫瘤[4]、肥胖和糖尿病[5]、神經退行性疾病[6]以及衰老[7]等疾病過程中可見JNK/SAPK作用的相關研究,然而關于JNK/SAPK在各種呼吸系統疾病中作用的研究非常少。在呼吸系統疾病中,有文獻報道,吸入含鹽酸的胃液可通過調控JNK/SAPK和ERK1/2從而阻止TNF-α受體基因轉錄而促進急性上呼吸道感染[8]。另有研究報道,結核桿菌通過調控JNK/SAPK和p38信號通路共同介導PI3K/PDK1/AKT信號通路,從而提高細胞趨化因子Leukotactin-1表達,加速肺結核形成[9]。除此之外,臨床研究報道,JNK/SAPK信號通路在呼吸道變態反應性疾病中起了至關重要作用[10]。然而其對慢性阻塞性肺疾病(COPD)的影響尚未明確。2015年5月~2016年3月,本研究構建大鼠COPD模型,采集肺組織標本,觀察COPD大鼠肺組織JNK/SAPK表達變化,分析JNK/SAPK與COPD發生發展的相關性。
1 材料與方法
1.1 材料 Ⅱ級Wistar雄性大鼠16只,6~7周齡,體質量(200±20)g,購于第二軍醫大學動物實驗中心,SPF級,適應性飼養一周。鼠代謝籠(L2890339,西域機電系統有限公司);勻漿機(德國IKA公司,型號:T25 basic);Buxco PFT系統肺功能檢測儀(美國BUXCO公司);臺式離心機(美國Eppendorf 公司);戊巴比妥鈉進口分裝(上海國藥集團);脂多糖(美國Sigma公司);一抗JNK/SAPK試劑盒和二抗(上海寶曼生物科技有限公司);TRIzol試劑(上海生工生物工程公司);RNA分離純化試劑盒(加拿大BBI公司)。
1.2 分組及處理 將大鼠隨機分為對照組和COPD組,各8只。對照組不予任何處理。COPD組采用煙霧熏吸法。將大鼠放入自制的有機玻璃染毒箱(100 cm×80 cm×50 cm)內,香煙點燃后,用60 mL注射器吸滿煙霧,然后通過三通管將香煙煙霧快速注入染毒箱內。為減少香煙燃燒所產的水蒸氣對大鼠的影響,在箱底放置適量硅膠干燥劑。每次持續時間大約1 h、間隔4 h一次,共28 d,同時第1、14天給予氣管內注入脂多糖。
1.3 肺組織形態學觀察 28 d后處死大鼠,暴露肺部,取部分左肺及氣管支氣管置于10%甲醛溶液中充分固定48 h。梯度乙醇脫水、二甲苯透明、浸蠟(時間>3 h),包埋,每一組織塊行厚4 μm連續切片兩張,常規HE染色,于光學顯微鏡下攝片觀察肺組織形態學改變。
1.4 肺功能的測定 將大鼠以4%的戊巴比妥鈉(50 mg/kg)腹腔注射麻醉,仰臥位固定于大鼠固定板,縱行切開頸部皮膚約2 cm,機械鈍性分離皮下組織,暴露并分離氣管后,在第三、四氣管環中間切開倒“T”型切口,將氣管插管插入氣管中,并用線系緊。動物肺功能分析系統調試后,將大鼠放入體描箱,將氣管插管與體描箱氣路相連。設定呼吸機參數:呼吸比15∶10,呼吸頻率75次/min。調整流量閥,使動物的潮氣量為5 mL/kg。從氣道壓力曲線上觀察動物是否有呼吸對抗。如有呼吸對抗,則逐次按50 mg/kg劑量追加麻醉,直至對抗消失。呼吸對抗消失后,關閉體描箱,點擊“開始”按鈕,開始測定肺功能,采集數據。
1.5 大鼠肺組織中TNF-α水平檢測 采用ELISA法。暴露肺部,取部分左肺組織,加入生理鹽水,勻漿。用大鼠TNF-α單抗包被于酶標板上,標準品與樣品中的TNF-α與單抗結合,加入生物素化的抗大鼠TNF-α形成免疫復合物連接在板上,辣根過氧化物酶標記的Streptavidin與生物素結合,加入底物工作液顯藍色,最后加終止液硫酸,在波長450 nm處測A值,TNF-α水平與A值成正比,可通過繪制標準曲線求出標本中TNF-α水平。
1.6 肺組織JNK/SAPK蛋白相對表達的檢測 采用免疫組化法。取部分右肺組織樣本浸泡于4%多聚甲醛中固定、包蠟、切片(厚度為4 μm),貼片于載玻片,60 ℃烤片,存于通風處。免疫組化法順序如下:脫蠟,抗原修復,加入一抗(靶蛋白),4 ℃過夜;次日,加入二抗、HRP生物標志鏈,DAB顯色,蘇木素染色,鹽酸乙醇分化,自來水沖洗泛藍20 min,常規脫水、透明、封片。采用Image J軟件對結果進行分析,鏡下觀察到棕黃色顆粒為陽性表達,每張切片取10個高倍鏡視野,以陽性細胞的積分吸光度(IA)為判定指標進行統計。
1.7 肺組織JNK/SAPK mRNA相對表達水平的檢測 采用RT-PCR技術。取部分右肺組織,以高速勻漿機制備組織勻漿。以TRIzol試劑進行總RNA提取,以Oligo DT為引物從總RNA中合成cDNA,然后用上述特異性引物進行PCR擴增。RT-PCR反應檢測JNK/SAPK mRNA表達,GAPDH作為內參。JNK上游引物: 5′GGTATGCCCAAGAGGACAGAGGA3′,下游引物:5′AGCCCAGATAGAGCCAGTCGTAA3′,產物228 bp。GAPDH 上游引物:5′CCTTCCGTGTTCCTACCCCC3′,下游引物:5′AGCCCAAGATGCCCTTCAGT 3′,產物481 bp。以系統的SPOT DENSITY軟件分別測量內參和JNK/SAPK mRNA基因擴增條帶的密度,以JNK/SAPK mRNA條帶的密度與內參基因的密度比值代表基因表達的相對水平。
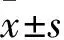
2 結果
2.1 兩組肺組織病理表現 對照組(圖1A)肺組織無明顯變化;COPD組(圖1B)小氣道上皮細胞變性壞死糜爛脫落,氣道黏膜充血水腫,同時管腔內可見較多中性粒細胞聚集和炎性滲出物,氣管、支氣管周圍大量炎性細胞浸潤,管腔變小、狹窄,肺泡管、肺泡囊及肺泡明顯擴大,部分肺泡出現融合現象。肺泡數量減少,肺泡毛細血管床減少,以上表現均與COPD的病理改變一致,說明造模成功(圖1)。

圖1 對照組 (A)和COPD組(B)大鼠肺組織病理變化
2.2 兩組肺功能指標比較 與對照組比較,COPD組大鼠第0.3秒用力呼氣容積與用力呼氣容積的比值(FEV0.3/FVC)降低,呼氣阻力(Ri)、吸氣阻力(Re)升高,動態順應性(Cldyn)下降(P<0.05或<0.01)。見表1。
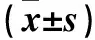
表1 兩組肺功能指標比較
注:與對照組比較,△P<0.05,△△P<0.01。
2.3 兩組肺組織TNF-α、JNK/SAPK蛋白、JNK/SAPK mRNA比較 與對照組比較,COPD組TNF-α、JNK/SAPK蛋白、JNK/SAPK mRNA表達水平升高(P均<0.05)。見表2。
表2 兩組肺組織TNF-α、JNK/SAPK蛋白、JNK/SAPK mRNA表達比較
注:與對照組比較,△P<0.05,△△P<0.01。
2.4 JNK/SAPK mRNA表達水平與TNF-α的相關性 JNK/SAPK mRNA表達水平與TNF-α呈正相關(r=0.751,P<0.05)。
3 討論
COPD是一種可防治的慢性氣道炎癥性疾病,吸煙為其主要誘因,炎癥反應為其發生發展的主要病理機制,病變部位主要涉及全部氣道、肺泡壁、肺部血管和肺實質,涉及的細胞主要有中性粒細胞、上皮細胞、巨噬細胞、淋巴細胞等,激活的炎癥細胞釋放多種炎癥介質和細胞因子,包括IL-8、IL-6、TNF-α、MMP-9、IL-1、GM-CSF等近50種細胞因子。研究發現,COPD患者的小葉中心肺氣腫部位、小氣道、肺泡巨噬細胞、肺泡灌洗液和血管有顆粒污染物殘留[11]。同時,香煙煙霧中含有丙烯醛、過氧化氫及活性氧等多種氧化物,能通過增加體內氧化負荷及活化炎性細胞,釋放內源性氧化劑,從而打破體內氧化/抗氧化系統平衡,引起肺部炎癥[12]。另外,煙霧還可誘導中性粒細胞釋放蛋白酶,導致蛋白酶-抗蛋白酶系統的失衡,損害肺組織彈力纖維,從而進一步導致肺氣腫形成。鑒于COPD與吸煙密切相關以及COPD發生發展與炎癥反應、氧化損傷、蛋白酶/抗蛋白酶失調、免疫障礙等多個方面異常密切相關,因此,近年對COPD治療研究逐步受到醫學界廣泛重視。
JNK/SAPK是一類絲氨酸/蘇氨酸激酶,為哺乳動物絲裂素活化蛋白激酶家族中的一員。JNK/SAPK被激活后,能通過不同的下游分子調控機體和細胞的各種功能,如mRNA轉錄、蛋白合成、線粒體功能等,還可以調控細胞自噬。JNK/SAPK信號通路是機體正常與疾病狀態時細胞調節的重要靶點。而JNK/SAPK磷酸化,可以在炎癥反應方面發揮免疫調節作用。
Oltmanns等[13]通過構建支氣管哮喘模型發現,JNK是支氣管哮喘發病過程中重要的信號分子,在氣道炎癥中發揮重要作用,可促進氣道平滑肌細胞分泌大量炎性因子如活性調節蛋白、粒單集落刺激因子、IL-8等。Bennett等[14]發現,JNK/SAPK的特異性抑制劑SP600125(一種可逆的ATP競爭性抑制劑)可抑制c-Jun的磷酸化及一些炎癥因子如TNF-α、IL-2、INF-γ的表達。而吸煙可直接或間接損傷氣道組織細胞,使肺局部中性粒細胞聚集、巨噬細胞凋亡,并激活、釋放多種炎性介質,如TNF-α、IL-2、IL-6、INF-γ、巨噬細胞炎性蛋白、粒單集落刺激因子和蛋白酶[15~17],產生異常炎癥反應,是COPD發病的中心環節。近年來,人們已意識到JNK/SAPK信號通路在氣道炎癥反應中可能具有重要意義。但有關JNK/SAPK蛋白磷酸化水平的變化及其與COPD炎癥反應機制的關系尚未見報道。
本研究發現,構建煙霧誘導的大鼠COPD模型,大鼠出現了精神萎靡,活動量減少,寒戰明顯,呼吸急促,呼吸動度增加,噴嚏較多,部分大鼠鼻部出現黏性分泌物,呼吸時伴有明顯痰鳴音,心率明顯加快;光鏡下出現了大量炎性細胞浸潤,肺泡壁變薄,部分肺泡融合,肺泡數量減少,肺泡毛細血管床減少等典型的COPD病理現象;肺功能提示FEV0.3/FVC明顯下降,Ri、Re升高,Cldyn下降,表明成功建立COPD大鼠模型。采集肺組織標本,分析JNK/SAPK在肺組織中的表達及其與COPD發展的相關性。免疫組化結果顯示,COPD大鼠肺組織中JNK/SAPK蛋白表達增強。與對照組相比,COPD組大鼠JNK/SAPK mRNA表達水平升高,差異有統計學意義,且與TNF-α有顯著的線性相關關系。考慮JNK/SAPK導致氣道炎癥和肺氣腫,促進COPD發生發展,與JNK/SAPK磷酸化導致一些炎癥因子如TNF-α、IL-2、INF-γ的表達升高有關。而肺組織的異常炎癥反應則是COPD發病的中心環節。因此JNK/SAPK在煙霧誘導的COPD發生發展中發揮了一定作用。根據以上體內實驗,我們推測,JNK/SAPK信號通路可能通過激活煙霧誘導的炎癥因子來導致氣道炎癥和肺氣腫發病。
綜上所述,本研究初步探討了JNK/SAPK在煙霧誘導COPD大鼠中的表達及作用,認為JNK/SAPK可以通過激活煙霧誘導的相關炎癥因子,促進氣道炎癥和肺氣腫,導致COPD發生。但其更加準確的調控機制還需進一步探討。同時,JNK/SAPK很有可能成為COPD治療的新靶點,而JNK/SAPK研究理論的完善,必將為未來COPD及其他呼吸系統疾病的治療提供新的思路。
[1] Takahashi O, Komaki R, Smith PD, et al. Combined MEK and VEGFR inhibition in orthotopic human lung cancer models results in enhanced inhibition of tumor angiogenesis, growth, and metastasis[J]. Clin Cancer Res, 2012,18(6):1641-1654.
[2] Zhou YY, Li Y, Jiang WQ, et al. MAPK/JNK signaling: a potential autophagy regulation pathway[J]. Biosci Rep, 2015,35(3) pii:e00199.
[3] Ryter SW, Nakahira K, Haspel JA, et al. Autophagy in pulmonary diseases[J]. Annu Rev Physiol, 2012,74(10):377-401.
[4] Tournier C. The 2 faces of JNK signaling in cancer[J]. Genes Cancer, 2013,4(9-10):397-400.
[5] Bennett BL, Satoh Y, Lewis AJ. JNK: a new therapeutic target for diabetes [J]. Curr Opin Pharmacol, 2003,3(4):420-425.
[6] Mehan S, Meena H, Sharma D, et al. JNK: a stress-activated protein kinase therapeutic strategies and involvement in Alzheimer′s and various neurodegenerative abnormalities[J]. J Mol Neurosci, 2011,43(3):376-390.
[7] Gabai VL, Meriin AB, Yaglom JA, et al. Role of Hsp70 in regulation of stress-kinase JNK: implications in apoptosis and aging[J]. FEBS Lett, 1998,438(1-2):1-4.
[8] Maniatis NA, Sfika A, Nikitopoulou I, et al. Acid-induced acute lung injury in mice is associated with p44/42 and c-Jun N-terminal kinase activation and requires the function of tumor necrosis factor α receptor I[J]. Shock, 2012,38(4):381-386.
[9] Cho JE, Park S, Cho SN, et al.c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (p38 MAPK) are involved in mycobacterium tuberculosis-induced expression of Leukotactin-1[J]. BMB Rep, 2012,45(10):583-588.
[10] Bennett BL. c-Jun N-terminal kinase-dependent mechanisms in respiratory disease[J]. Eur Respir J, 2006,28(3):651-661.
[11] Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease[J]. N Engl J Med, 2004,350(26):2645-2653.
[12] Bodas M, Tran I, Vij N. Therapeutic strategies to correct proteostasis-imbalance in chronic obstructive lung diseases[J]. Curr Mol Med, 2012,12(7):807-814.
[13] Oltmanns U, Issa R, Sukkar MB, et al. Role of c-jun N-terminal kinase in the induced release of GM-CSF,RANTES and IL-8 from human airway smooth muscle cells[J]. Br J Pharmacol, 2003,139(6):1228-1234.
[14] Bennett BL, Sasaki DT, Murray BW, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal Kinase[J]. Proc Natl Acad Sci USA, 2001,98(24):13681-13686.
[15] Provinciali M, Cardelli M, Marchegiani F. Inflammation, chronic obstructive pulmonary disease and aging[J]. Curr Opin Pulm Med, 2011,17(Suppl 1):3-10.
[16] Kim V, Rogers TJ, Criner GJ. New concepts in the pathobiology of chronic obstructive pulmonary disease[J]. Proc Am Thorac Soc, 2008,5(4):478-485.
[17] Murugan V, Peck MJ. Signal transduction pathways linking the activation of alveolar macrophages with the recruitment of neutrophils to lungs in chronic obstructive pulmonary disease[J]. Exp Lung Res, 2009,35(6):439-485.
Changes in levels of JNK/SAPK of rats with chronic obstructive pulmonary disease
TIANXue,ZHANGXingyi,LIFeng,XIAOHui,ZHOUXin
(ShanghaiGeneralHospitalofShanghaiJiaotongUniversity,Shanghai200080,China)
Objective To investigate the changes in levels of c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) in the lung tissues of rats with chronic obstructive pulmonary disease (COPD). Methods Sixteen Ⅱ male Wistar rats were randomly divided into two groups: the control group and COPD group. The control group was not treated, while the COPD group was stimulated with smoke to set up COPD models. After models were established, we detected the lung pathology through optical microscope, pulmonary function through animal pulmonary instrument, TNF-α expression in lung tissue through ELISA, JNK/SAPK expression through immunohistochemistry, and JNK mRNA expression through RT-PCR. Results In the COPD group, light microscope showed that there was a large number of inflammatory cell infiltration, alveolar fusion, and alveolar reduction, which proved the COPD model was successful. Compared with the control group, FEV0.3/FVC decreased, Ri and Re increased, and Cldyn decreased (P<0.05 orP<0.01), and TNF-α increased in the COPD group (P<0.05). Immunohistochemistry showed that JNK/SAPK expression improved in the COPD group. RT-PCR result showed JNK mRNA expression increased in the COPD group as compared with that of the control group (allP<0.05). JNK mRNA expression was positively correlated with TNF-α (r=0.751,P<0.05).Conclusion The level of JNK/SAPK increases in smoke-stimulated COPD rats, which can cause airway inflammation and emphysema, and promote the occurrence of COPD through activating smoke-induced inflammatory cytokines.
chronic obstructive pulmonary disease; c-Jun N-terminal kinase; stress-activated protein kinase; effect mechanism
國家自然科學基金面上項目(81470218);中國醫師協會呼吸免疫專項課題(2014)。
田雪(1984-),女,主治醫師,主要研究方向為慢性阻塞性肺疾病的發病機制。E-mail:lwtianxue@126.com
周新(1953-),男,主任醫師,主要研究方向為慢性阻塞性肺疾病的發病機制。E-mail:xzhou53@163.com
10.3969/j.issn.1002-266X.2016.45.005
R563.13
A
1002-266X(2016)45-0015-04
2016-09-11)
