重抽樣方差成分檢驗的多位點關聯(lián)分析*
2017-01-10 03:46:33唐明生黃水平金英良張耀東趙楊陳峰曾
中國衛(wèi)生統(tǒng)計 2016年6期
唐明生黃水平金英良張耀東趙 楊陳 峰曾 平△
重抽樣方差成分檢驗的多位點關聯(lián)分析*
唐明生1黃水平1金英良1張耀東1趙 楊2陳 峰2曾 平1△
目的在線性混合模型框架下探索基于重抽樣方差成分的似然比檢驗多位點關聯(lián)分析方法。方法假設SNP位點效應服從正態(tài)分布,將多位點關聯(lián)分析轉化為對隨機效應方差的似然比檢驗,采用重抽樣方法獲得統(tǒng)計量的零分布,通過混合分布提高計算速度。結果模擬研究顯示重抽樣檢驗以及混合分布表現(xiàn)良好,能夠有效控制I型錯誤,統(tǒng)計效能優(yōu)于現(xiàn)有方法。結論似然比檢驗是一種高效能的多位點關聯(lián)檢驗方法,通過對置換和bootstrap分布的有效近似,基于重抽樣的似然比方差成分檢驗,在保持良好統(tǒng)計效能的同時,能明顯降低計算負荷。
關聯(lián)研究 方差成分 線性混合模型 似然比檢驗 置換法 非參數(shù)bootstrap法
在全基因組關聯(lián)性研究(genome w ide association study,GWAS)中,相對于單位點分析,多位點分析已經(jīng)成為一種有效的關聯(lián)性檢驗方法[1-8]。單位點分析對成千上萬個單核苷酸多態(tài)性(single nucleotide polymorphisms,SNP)依次檢驗,要求十分嚴格的多重檢驗水準[9]。相反,多位點分析同時檢驗一組SNP是否與表型相關,有諸多優(yōu)點:能顯著降低多重比較負擔,能利用SNP間連鎖不平衡信息來提高效能。考慮到位點間聯(lián)合效應,SNP集合常根據(jù)基因選定,也即位于相同基因內的變異位點組成一個集合[10]。
多位點分析可采用固定效應模型和固定效應檢驗執(zhí)行,如Wald檢驗;但固定效應檢驗在SNP位點個數(shù)較多時效能低下。另一方面,在線性混合模型下假設SNP具有隨機效應,能夠將多位點分析轉化為對隨機效應的方差成分檢驗;現(xiàn)階段主要通過得分檢驗來實現(xiàn)這一目的[1,8,11-16]。得分檢驗只需擬合零模型,計算效率高;但結果相對保守,特別是在小樣本的情況下[16]。本文嘗試采用似然比檢驗(likelihood ratio test,LRT)及限制性似然比檢驗(restricted likelihood ratio test,ReLRT)對SNP集合進行關聯(lián)性檢驗[17-19],通過置換及bootstrap[20-23]等重抽樣方法獲得統(tǒng)計量的零分布。事實上,在關聯(lián)性研究中為了避免導出解析解,重抽樣技術已經(jīng)廣泛應用于復雜假設檢驗[6,24-29]。然而,重抽樣屬于計算密集型統(tǒng)計方法,為了減少計算負荷,本文進一步通過混合分布來近似重抽樣分布,以提高似然比檢驗的計算速度。最后通過模擬和實際數(shù)據(jù)來展示本文的方法。
資料與方法
1.線性混合模型和似然的推斷
用線性混合模型[30-32]來描述多個SNP位點和表型之間的關系:

Y是n維連續(xù)性表型,X為n×m的協(xié)變量矩陣(m為協(xié)變量個數(shù)),具有固定效應α,G表示n×p的SNP基因型矩陣(編碼0,1,2,p為SNP個數(shù)),具有隨機效應b,服從均數(shù)為0、方差為τ的正態(tài)分布。ε為殘差項,服從均數(shù)為0、方差為σ2的正態(tài)分布。
檢驗G與表型Y之間的關系等價于檢驗隨機效應H0:b=0,也等價于檢驗方差成分H0:τ=0。這樣,通過混合效應模型SNP集合的關聯(lián)分析就轉化為對隨機效應方差成分的假設檢驗。
省略常數(shù)項,模型(1)的對數(shù)似然函數(shù)和限制性對數(shù)似然函數(shù)分別為:
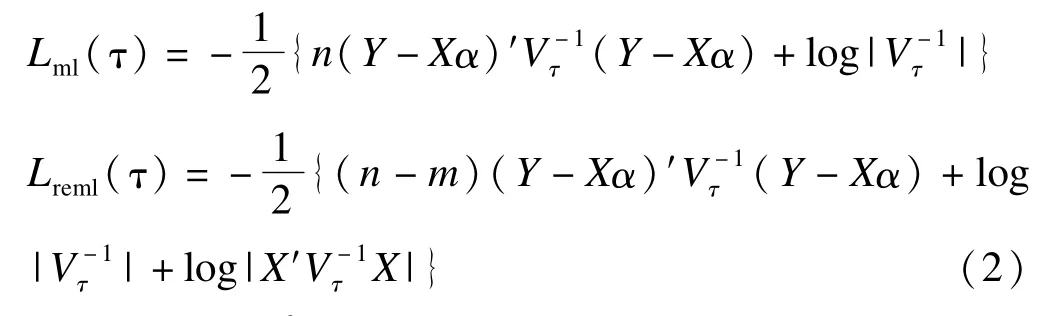

與得分檢驗不同,似然比檢驗和限制性似然比要求同時擬合零模型和備擇模型,本文通過R軟件的lm和lme軟件包[33-34]來估計線性模型和混合效應模型。
在H0的條件下,τ位于參數(shù)空間的邊緣;由于這種限制,TLRT和TReLRT的零分布不再漸近服從標準的卡方分布[35],先前的研究顯示在一定條件下,TLRT和TReLRT漸近服從的混合分布[18]。但是,該混合分布的假定條件在實際中很難得到滿足。Pinheiro和Bates提出了0.65:0.35的混合分布[36]。本文模擬研究表明,在多位點似然比關聯(lián)性分析中兩種混合比例都不正確。事實上,混合分布比例參數(shù)依具體情況而定,依賴于特殊矩陣的特征值[37-39]。本文通過重抽樣方法來獲得似然比統(tǒng)計量和限制性似然比統(tǒng)計量的零分布。
2.重抽樣算法
當烘絲機入料電子秤不再檢測到煙絲,烘絲機即由生產(chǎn)狀態(tài)切換至收尾狀態(tài),筒壁Ⅰ區(qū)、Ⅱ區(qū)溫度控制器停止工作,Ⅰ區(qū)、Ⅱ區(qū)蒸汽壓力控制閥保持其最后適用的修正變量60 s后;Ⅰ區(qū)、Ⅱ區(qū)蒸汽壓力控制閥開始逐漸關閉Ⅰ區(qū)、Ⅱ區(qū)蒸汽閥門開度,在20 s內完全關閉Ⅰ區(qū)、Ⅱ區(qū),蒸汽停止加熱筒壁。在此過程中,因為烘絲機蒸汽壓力控制閥對筒壁Ⅰ區(qū)、Ⅱ區(qū)蒸汽壓力保持以及對Ⅰ區(qū)、Ⅱ區(qū)蒸汽的關閉均為同時進行,且烘絲機由收尾狀態(tài)至蒸汽壓力保持的延時時間以及對筒壁Ⅰ區(qū)和Ⅱ區(qū)蒸汽壓力的保持時間較長,使烘絲機熱慣性過大,導致了收尾狀態(tài)產(chǎn)生大量的干尾煙絲。
置換和bootstrap法都是通過重復抽樣獲得零分布,不同之處在于兩者的抽樣方式。置換法通過擾亂原始數(shù)據(jù)的標簽產(chǎn)生,需要將Y和X視為一個整體,即置換是對Y和X同時進行,而保持G固定不變;這樣可以保持Y與X的相關結構。這里潛在假設G和X是獨立的。bootstrap法采用非參數(shù)有放回抽樣產(chǎn)生,即是從中有放回抽得的樣本。以上兩種產(chǎn)生數(shù)據(jù)D*的方法是等價的[21-22]。在bootstrap算法中,最自然的抽樣是從原始殘差X中進行抽樣;然而Davison和Hinkley[21]指出,當是異方差性時,從修正殘差中 抽樣更好,h為杠桿值。置換和bootstrap檢驗的P值采用蒙特卡洛方法獲得,即通過有限次數(shù)(設為S)的重抽樣方式代替。很多研究表明,在檢驗水準較大時(如0.05),S=1000或2000時P值就趨于相對穩(wěn)定[20-22]。
3.近似分布
假設TLRT和TReLRT服從混合分布
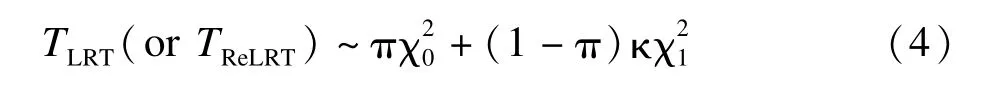
κ是未知的尺度參數(shù)分別為自由度為0和1的卡方分布。假設近似零分布為卡方混合的原因在于:(I)在適當?shù)臈l件下,TLRT和TReLRT取0的概率不為零,因此包括作為退化到0的分布;(II)在標準條件下,即參數(shù)在其參數(shù)空間的內部時,TLRT和TReLRT漸近服從自由度為1的卡方分布[40],因此包括表示可能的偏離;類似的混合分布也被應用在其他地方[17,36]。κ的估計值為:

本文采用局部概率法來估計π[38-39]:
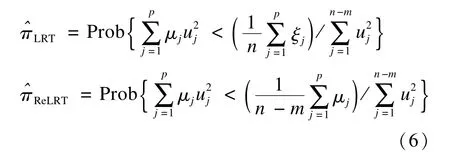
μ是G′P0G的特征值,P0=In-X(X′X)-1X′,ξ是G′G的特征值,u是獨立標準正態(tài)分布的隨機變量。局部概率法相對于矩估計法的優(yōu)勢在于:其π估計值與L的選擇無關,因此π估計值是穩(wěn)定的,并會導致更加精確的估計。對(6)式可采用Davies法[42]獲得π的估計值然后將代入(5)中可算得式(6)還進一步表明:TLRT和TReLRT的零分布依具體情況而定,固定比例的卡方混合分布是不恰當?shù)摹?/p>
結 果
用FEV1下降率的數(shù)據(jù)[43]來展示本文提出的LRT和ReLRT多位點關聯(lián)性分析方法,樣本量為301,用FEV1下降的斜率ES作為表型[43]。結果表明,位點rs9469089與FEV1相關聯(lián);該位點位于染色體6p21.32,在基因RNF5的第一個內含子區(qū)域內,而RNF5編碼膜結合型泛素連接酶。在該數(shù)據(jù)中RNF5一共包含14個SNP。
首先評價I類錯誤控制。假設表型來自于

X1和X2分別為標準正態(tài)變量和二分類變量,模擬次數(shù)為105次。重抽樣中S=2000,對于近似混合分布,取L=2000、1500、1000、800、500、300和100。用m lp1-m lp7對應以上各L取值。在評價統(tǒng)計效能時,假設表型來自于
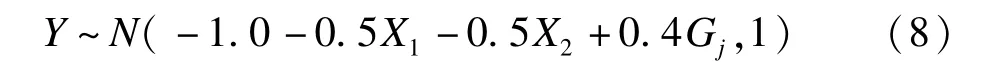
這里,j取1到14,即每個SNP位點依次被設為關聯(lián)位點,效應值為0.20,0.30和0.40,重復103次,則一共運行1.4×104次。P值用小于檢驗水準α=0.05的比例估計。
2.解釋和說明
模擬結果顯示,在LRT和ReLRT中κ與π的平均值分別為1.118和0.909與0.865和0.604;這些數(shù)值和本文特定的基因型矩陣和協(xié)變量矩陣相關,其他不同的選擇會導致不同的κ和π估計值。這表明Self和Liang[17]及Pinheiro和Bates[36]提出的零分布是不合理的,同時也表明LRT和ReLRT服從不同的零分布。圖1表明參數(shù)π在有限的范圍內變化(這是因為在所有的模擬中使用相同的G和相似的X),將其固定取某一常數(shù)是不適當?shù)摹8鶕?jù)公式(6),π隨數(shù)據(jù)集而變化,因此直接根據(jù)數(shù)據(jù)估計π更合理。此外,模擬還表明,相對于在ReLRT中,κ估計值在LRT中更大(圖1)。κ估計值的精度隨著L減小而降低;然而,不同L值通常產(chǎn)生非常相似的結果。
圖2表明對LRT而言,模擬算法[39,45]估計的I類錯誤率結果偏于保守,而置換和bootstrap檢驗可以調整LRT的這種保守性;混合分布對I類錯誤的控制與L取值無關。ReLRT在所有情形下對I類錯誤控制都表現(xiàn)良好;這可能是因為LRT對方差成分的估計是有偏估計,而ReLRT對方差成分的估計是無偏估計[46-47]。LRT模擬算法的統(tǒng)計效能低于相應置換和bootstrap檢驗的統(tǒng)計效能;在ReLRT中,模擬算法、置換和bootstrap檢驗的統(tǒng)計效差異微小(圖3)。此外,通常次要等位基因頻率高,統(tǒng)計效能也較高(圖3)。
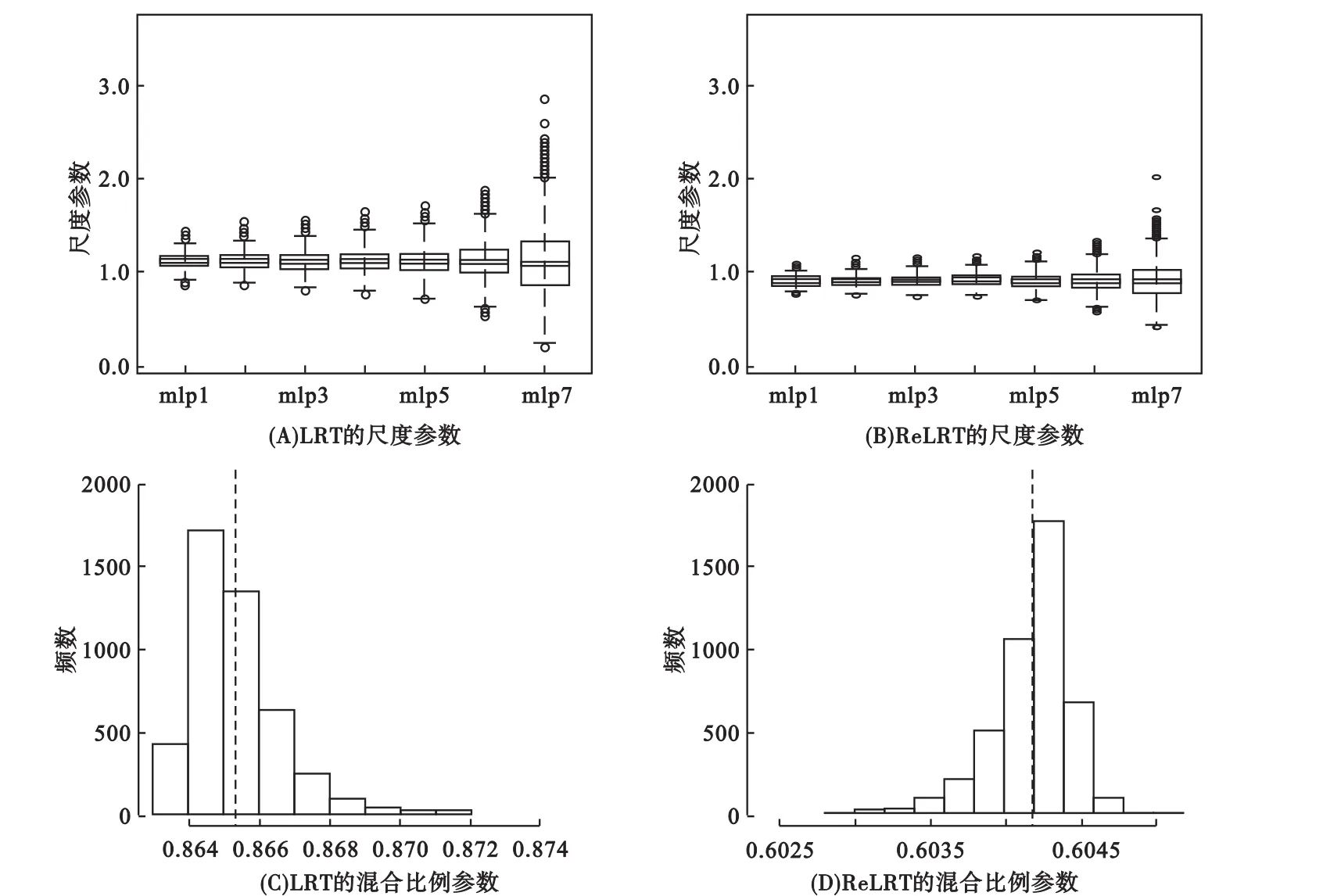
圖1 在不同的L值下,LRT和ReLRT近似混合分布的尺度參數(shù)κ和混合比例參數(shù)π的估計值
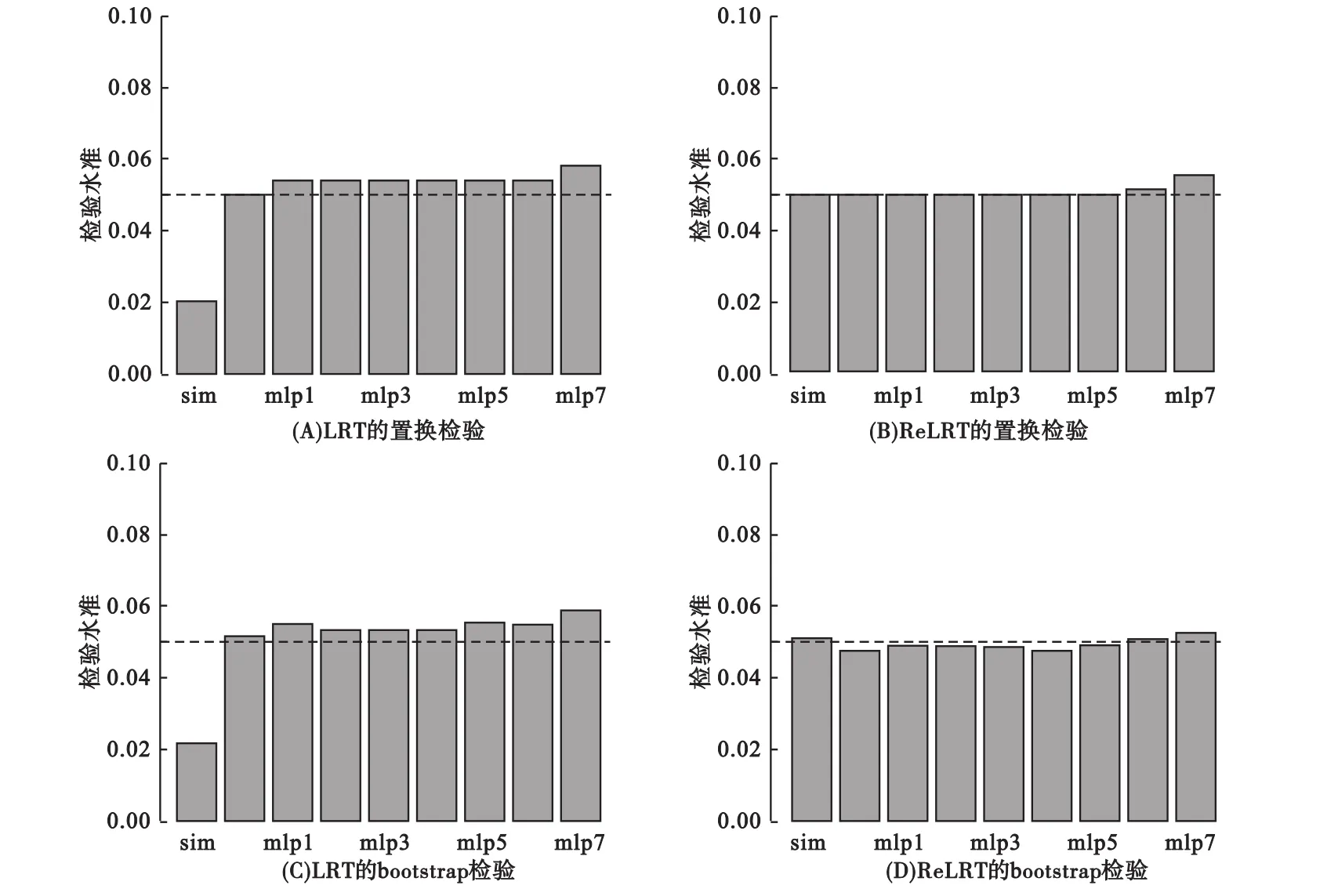
圖2 LRT和ReLRT I類錯誤率的置換或bootstrap算法估計
LRT和ReLRT的統(tǒng)計效能通常高于得分檢驗的統(tǒng)計效能。對于得分檢驗、LRT和ReLRT,在模擬效應為0.40時三者的平均統(tǒng)計效能分別為0.474、0.544和0.545;模擬效應為0.30時為0.315,0.453和0.510;模擬效應為0.20時為0.179,0.232和0.250。圖3表明:對于LRT(或ReLRT),置換和bootstrap兩種檢驗方法的統(tǒng)計效能相似。
在FEV1數(shù)據(jù)中將斜率(即ES)作為表型[43],內毒素暴露和BMI作為協(xié)變量,采用LRT和ReLRT以及得分檢驗來分析基因RNF5的關聯(lián)性(表1)。從表1可見,在LRT中由模擬算法獲得的P值比其他情形下的P值大,置換和bootstrap檢驗的P值更小;ReLRT的P值變化較小。這些結果和模擬的結論一致。得分檢驗的P值為0.027。
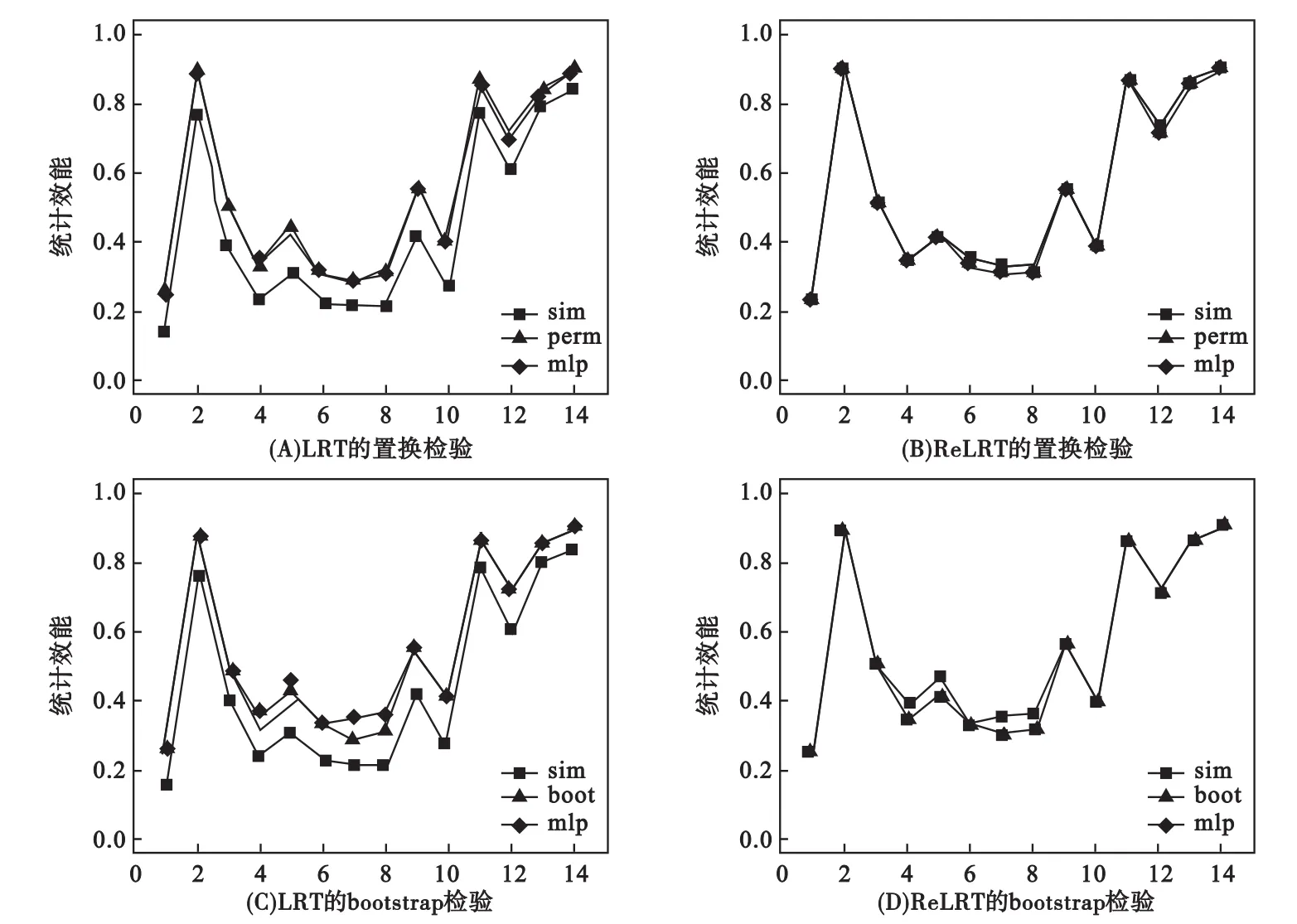
圖3 LRT和ReLRT統(tǒng)計效能的置換或bootstrap算法估計
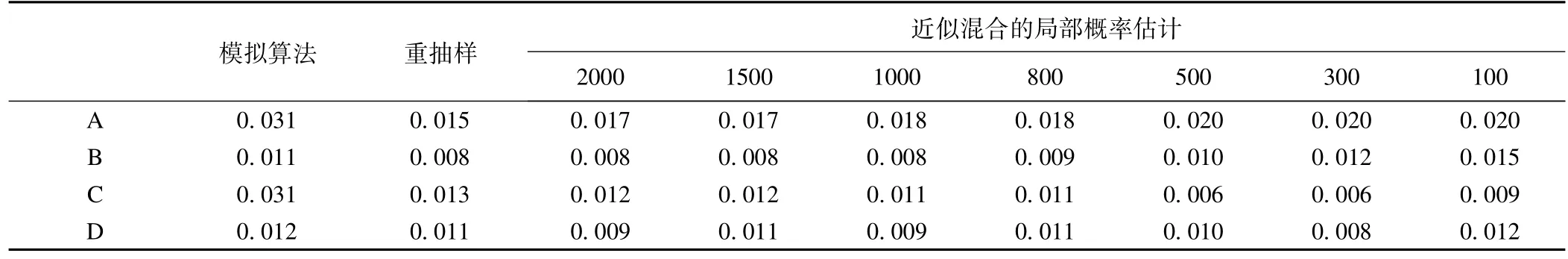
表1 FEV1數(shù)據(jù)中基因RNF5的P值
討 論
本文在線性混合模型框架下研究了基于重抽樣的似然比多位點關聯(lián)分析,在該框架下,一組SNP位點與表型的關聯(lián)性分析被轉化為對隨機效應的方差成分檢驗[1,11-16]。本文采用重抽樣技術(置換和bootstrap)獲得似然比統(tǒng)計量的零分布,避免了復雜的數(shù)學推導。此外,還通過混合分布近似置換或bootstrap分布。本文采用非參數(shù)而不是參數(shù)的bootstrap法,其原因在于非參數(shù)bootstrap法更加穩(wěn)健[20-21]。模擬表明,對于限制似然比檢驗,置換和bootstrap法能有效控制I類錯誤且統(tǒng)計效能高于現(xiàn)有的得分檢驗方法;然而,對于似然比檢驗,其I類錯誤未能得到有效控制。研究還發(fā)現(xiàn),對于小樣本基于模擬算法的似然比檢驗[39,41]會導致保守的結果,而隨著樣本量的增加,似然比檢驗表現(xiàn)趨于正常[45]。
重抽樣方法的主要缺點是耗時,本文利用混合分布近似來減少計算負荷。模擬表明,近似分布能顯著減少計算時間。例如,L=100時的混合分布比S=2000時的重抽樣檢驗計算時間大約減少20倍。更重要的是,在大規(guī)模多位點關聯(lián)分析時,要求更加精確的P值,這需要成千上萬次的置換或bootstrap抽樣,導致重抽樣方法不可行;而近似混合分布能夠適合這種情形。如前所述,在混合分布的參數(shù)估計中,和其他方法相比,局部概率法具有數(shù)值穩(wěn)定的優(yōu)點。
類似的重抽樣方法也被應用在其他情形,例如,F(xiàn)araway[48]和Samuh等[49]提出了線性混合模型似然比檢驗的參數(shù)bootstrap法,F(xiàn)itzmaurice等[50],Lee和Braun[51],Samuh[49]提出了似然比檢驗置換法,Sinha[52]在廣義線性混合模型下通過參數(shù)bootstrap法建立了方差成分的得分檢驗。本文的研究與上述文獻的區(qū)別在于:(I)上述研究主要針對的是縱向數(shù)據(jù),且這些數(shù)據(jù)在各水平內是相關的,而模型(1)事實上應用在看上去獨立的基于總體人群設計的遺傳數(shù)據(jù);(II)對于本文的似然比檢驗,同時采用置換和bootstrap法,且采用相對穩(wěn)健的非參數(shù)bootstrap法而不是參數(shù)bootstrap法;(III)將基于重抽樣的方法與其他方法(基于模擬的算法)進行比較[39],結果表明重抽樣方法更有效,雖然計算負荷更重;(IV)采用了有效的近似方法來提高計算效率。最后,有待進一步研究將其他方法(如,基于參數(shù)bootstrap的得分檢驗[52])運用到本文的研究中并進行比較。
[1]Wu MC,Kraft P,Epstein MP,et al.Powerful SNP-Set Analysis for Case-Control Genome-wide Association Studies.American Journal of Human Genetics,2010,86(6):929-942.
[2]Cai T,Lin X,Carroll RJ.Identifying genetic marker sets associated w ith phenotypes via an efficient adaptive score test.Biostatistics,2012,13(4):776-790.
[3]Maity A,Sullivan PF,Tzeng Ji.Multivariate Phenotype Association Analysis by Marker-Set Kernel Machine Regression.Genetic Epidem iology,2012,36(7):686-695.
[4]Schifano ED,Epstein MP,Bielak LF,et al.SNP Set Association A-nalysis for Fam ilial Data.Genetic Epidem iology,2012,36(8):797-810.
[5]Dai H,Zhao Y,Qian C,et al.Weighted SNP Set Analysis in Genome-W ide Association Study.PLoSONE,2013,8(9):e75897.
[6]Huang YT,Lin X.Gene set analysis using variance component tests.BMC Bioinformatics,2013,14(1):210.
[7]Jiao S,Hsu L,Bézieau S,et al.SBERIA:Set-Based Gene-Environment Interaction Test for Rare and Common Variants in Complex Diseases.Genetic Epidem iology,2013,37(5):452-464.
[8]Wu MC,Maity A,Lee S,etal.KernelMachine SNP-Set Testing Under Multiple Candidate Kernels.Genetic Epidemiology,2013,37(3):267-275.
[9]Balding D.A tutorial on statisticalmethods for population association studies.Nature Reviews.Genetics,2006,7(10):781-791.
[10]Prescott NJ,Lehne B,Stone K,et al.Pooled Sequencing of 531 Genes in Inflammatory Bowel Disease Identifies an Associated Rare Variant in BTNL2 and Implicates Other Immune Related Genes.PLoSGenetics,2015,11(2):e1004955.
[11]Liu D,Lin X,Ghosh D.Semiparametric Regression of Multidimensional Genetic Pathway Data:Least-Squares Kernel Machines and Linear M ixed Models.Biometrics,2007,63(4):1079-1088.
[12]Tzeng J,Zhang D.Haplotype-based association analysis via variancecomponents score test.American Journal of Human Genetics,2007,81(5):927-938.
[13]Kwee L,Liu D,Lin X,etal.A Powerful and Flexible Multilocus Association Test for Quantitative Traits.American Journal of Human Genetics,2008,82(2):386-397.
[14]Liu D,Ghosh D,Lin X.Estimation and testing for the effect of a genetic pathway on a disease outcome using logistic kernelmachine regression via logistic m ixed models.BMC Bioinformatics,2008,9(1):292.
[15]Tzeng J,Zhang D,Chang SM,et al.Gene-trait sim ilarity regression formultimarker-based association analysis.Biometrics,2009,65(3):822-832.
[16]Wu MC,Lee S,Cai T,etal.Rare-Variant Association Testing for Sequencing Data with the Sequence Kernel Association Test.American Journal of Human Genetics,2011,89(1):82-93.
[17]Self SG,Liang KY.Asymptotic Properties of Maximum Likelihood Estimators and Likelihood Ratio Tests under Nonstandard Conditions.JAM STAT ASSOC,1987,82(398):605-610.
[18]Stram DO,Lee JW.Variance Components Testing in the Longitudinal M ixed Effects Model.Biometrics,1994,50(4):1171-1177.
[19]Liang KY,Self SG.On the Asymptotic Behaviour of the Pseudolikelihood Ratio Test Statistic.JRoy Stat Soc,1996,58(4):785-796.
[20]Efron B,Tibshirani R.An Introduction to the bootstrap.New York:Chapman and Hall,1993.
[21]Davison AC,Hinkley DV.bootstrap Methods and Their Application.Cambridge:Cambridge University Press,1997.
[22]Good P.Permutation,Parametric,and bootstrap Tests of Hypotheses.3 edition.New York:Springer,2005.
[23]Efron B.The Jackknife,the bootstrap and Other Resampling Plans.Philadelphia:Society for industrial and applied mathematics,1982.
[24]Han F,Pan W.A data-adaptive sum test for disease association w ith multiple common or rare variants.Human Heredity,2010,70:42-54.
[25]Lin D.An efficientMonte Carlo approach to assesssing statistical significance in genom ic studies.Bioinformatics,2005,21(6):781-787.
[26]Madsen BE,Browning SR.A Groupw ise Association Test for Rare Mutations Using a Weighted Sum Statistic.PLoS Genetics,2009,5(2):e1000384.
[27]Lee S,Emond MJ,Bamshad MJ,et al.Optimal Unified Approach for Rare-Variant Association Testing w ith Application to Small-Sample Case-ControlWhole-Exome Sequencing Studies.American Journal of Human Genetics,2012,91(2):224-237.
[28]Ferkingstad E,Holden L,Sandve GK.Monte Carlo nullmodels for genom ic data.Statistical Science,2015,30(1):59-71.
[29]Lin D,Tang Z.A General Framework for Detecting Disease Associations w ith Rare Variants in Sequencing Studies.American Journal of Human Genetics,2011,89(3):354-367.
[30]Laird NM,Ware JH.Random-effects models for longitudinal data.Biometrics,1982,38(4):963-974.
[31]Littel R,M illiken GA,Stroup WW,et al.SAS for mixed models.SAS Institute Inc.Cary,NC,2006.
[32]Verbeke G,MolenberghsG.Linearmixedmodels for longitudinal data.New York:Springer,2009.
[33]Pinheiro J,Bates D,DebRoy S,et al.nlme:Linear and Nonlinear M ixed Effects Models.R package version 3.1-113.2013.
[34]R Core Team.R:A language and environment for statistical computing.R Foundation for Statistical Computing,Vienna,Austria.URL http://www.R-project.org/,2014.
[35]Molenberghs G,Verbeke G.Likelihood Ratio,Score,and Wald Tests in a Constrained Parameter Space.Am Stat,2007,61(1):22-27.
[36]Pinheiro JC,BatesD.M ixed-EffectsModels in S and S-PLUS,2nd edition.New York:Springer,2009.
[37]Kuo BS.Asymptotics of ML estimator for regression models with a stochastic trend component.Economet Theor,1999,15(1):24-49.
[38]Claeskens G.Restricted likelihood ratio lack-of-fit tests using mixed splinemodels.JR Stat Soc B,2004,66(4):909-926.
[39]Crainiceanu CM,Ruppert D.Likelihood ratio tests in linear mixed modelswith one variance component.J Roy Stat Soc B,2004,66(1):165-185.
[40]Lehmann EL,Romano JP.Testing statistical hypotheses,Third edition.New York:Springer,2006.
[41]Greven S,Crainiceanu CM,Küchenhoff H,et al.Restricted Likelihood Ratio Testing for Zero Variance Components in Linear M ixed Models.JComput Graph Stat,2008,17(4):870-891.
[42]Davies RB.Algorithm AS 155:The Distribution of a Linear Combination of chi-2 Random Variables.JR Stat Soc C,1980,29(3):323-333.
[43]Zhang R,Zhao Y,Chu M,et al.A Large Scale Gene-Centric Association Study of Lung Function in New ly-Hired Female Cotton Textile Workers w ith Endotoxin Exposure.PLoSONE,2013,8(3):e59035.
[44]Barrett JC,F(xiàn)ry B,Maller J,et al.Haploview:analysis and visualization of LD and haplotype maps.Bioinformatics,2005,21(2):263-265.
[45]Zeng P,Zhao Y,Liu J,et al.Likelihood Ratio Tests in Rare Variant Detection for Continuous Phenotypes.Annals of Human Genetics,2014,78(5):320-332.
[46]Patterson HD,Thompson R.Recovery of interblock information when block sizes are unqual.Biometrika,1971,58(3):545-555.
[47]Harville DA.Maximum Likelihood Approaches to Variance Component Estimation and to Related Problems.JR Stat Soc B,1977,72(358):320-338.
[48]Faraway JJ.Extending the linear model w ith R:generalized linear,m ixed effects and nonparametric regressionmodels.New York:Chapman&Hall/CRC,2005.
[49]Samuh MH,Grilli L,Rampichini C,et al.The Use of Permutation Tests for Variance Components in Linear M ixed Models.Commun Stat-Theor M,2012,41(16-17):3020-3029.
[50]Fitzmaurice GM,Lipsitz SR,Ibrahim JG.A Note on Permutation Tests for Variance Components in Multilevel Generalized Linear M ixed Models.Biometrics,2007,63(3):942-946.
[51]Lee OE,Braun TM.Permutation Tests for Random Effects in Linear M ixed Models.Biometrics,2012,68(2):486-493.
[52]Sinha SK.Bootstrap tests for variance components in generalized linearm ixed models.Can JStat,2009,37(2):219-234.
(責任編輯:郭海強)
國家自然科學基金項目(81402765);國家統(tǒng)計局統(tǒng)計科學研究項目(2014LY112)
1.徐州醫(yī)科大學公共衛(wèi)生學院流行病與衛(wèi)生統(tǒng)計學教研室(221004)
2.南京醫(yī)科大學公共衛(wèi)生學院生物統(tǒng)計學系
△通信作者:曾平,E-mail:zpstat@xzhmu.edu.cn
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
核科學與工程(2021年4期)2022-01-12 06:30:26
今日農(nóng)業(yè)(2020年19期)2020-12-14 14:16:52
中學生數(shù)理化·七年級數(shù)學人教版(2020年10期)2020-11-26 08:24:50
數(shù)學物理學報(2020年2期)2020-06-02 11:29:24
中學物理·高中(2016年12期)2017-04-22 11:53:03
光學精密工程(2016年6期)2016-11-07 09:07:19
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
