V2O5/γ-Al2O3表面酸性和釩價態的調控及其異丁烷脫氫性能
2017-01-21 02:19:52徐華龍
石油化工 2016年9期
張 林,秦 楓,黃 鎮,沈 偉,徐華龍
(復旦大學 化學系 上海市分子催化與功能材料重點實驗室 復旦大學先進材料實驗室,上海 200433)
V2O5/γ-Al2O3表面酸性和釩價態的調控
及其異丁烷脫氫性能
張 林,秦 楓,黃 鎮,沈 偉,徐華龍
(復旦大學 化學系 上海市分子催化與功能材料重點實驗室 復旦大學先進材料實驗室,上海 200433)
以γ-Al2O3為載體,采用等體積浸漬法制備了負載高分散氧化釩催化劑(V2O5/γ-Al2O3),并通過K,Ca,Mg對V2O5/ γ-Al2O3的表面酸性及釩價態進行調控,研究催化劑表面酸性改性的變化規律及與異丁烷脫氫活性和選擇性之間的構效關系。采用XRD、XPS、N2吸附-脫附、NH3-TPD和H2-TPR等方法對催化劑進行表征。實驗結果表明,當氧化釩負載量低于7%(w),氧化釩在氧化鋁表面處于良好分散狀態時,催化劑保持最佳的反應活性和選擇性。表征結果顯示,堿金屬K對V2O5/ γ-Al2O3表面的酸性和V的還原價態具有顯著的調節作用,當K添加量(w)為1.5%時,強酸中心幾乎被全部抑制,同時保留了適量的弱酸中心,而V的還原價態由2.87+調至3.95+;在1.5%K-V2O5/γ-Al2O3催化劑上異丁烷脫氫反應340 min后,異丁烷轉化率保持在30%以上,異丁烯選擇性保持在97%以上。
V2O5/γ-Al2O3催化劑;表面酸性;釩價態;異丁烷脫氫
低碳烷烴催化脫氫合成低碳烯烴是化石能源高值轉化的重要路徑之一,高效和穩定催化劑的研發是該工藝路線成功的關鍵點。目前,異丁烷脫氫主要采用Pt-Sn或Cr2O3/Al2O3催化劑,載體的表面酸中心位對催化活性起重要作用,反應溫度通常在555~649 ℃[1-4],在該反應溫度下脫氫產物異丁烯非常容易在強酸性位發生異構化、裂解、芳構化和聚合等副反應,導致催化劑的活性和選擇性下降[5]。為了抑制這類現象發生,工業中采取了添加堿金屬來調變催化劑表面酸性的措施,如在FBD-4工藝中,在鉻催化劑中添加約2%(w)的K;在Oleflex工藝中,將Cs摻雜在Pt-Sn催化劑中[4]。但由于Cr和Pt基催化劑存在環境和經濟性問題,在工業化應用中將會受到很大制約。
近年來,釩基催化劑應用于低碳烷烴脫氫和氧化脫氫合成烯烴的研究受到廣泛關注。氧化脫氫具有轉化率高和反應溫度低的特點,但氧化反應深度不易控制,導致異丁烯的選擇性較低[5]。Ogonowski等[6-7]發現在V2O5/Al2O3催化劑上,用二氧化碳作為溫和氧化劑時,仍會導致異丁烯選擇性的下降。相比較而言,低碳烷烴直接脫氫具有更好的選擇性。Volpe等[8]通過對比了γ-Al2O3,α-Al2O3和USY沸石等載體,發現載體的酸性對脫氫的轉化率和選擇性具有顯著影響。Harlin等[9]研究結果發現,酸性載體上釩催化劑的反應活性與氧化釩在表面的分散狀態和價態有關,V3+和V4+均是異丁烷脫氫的活性中心。而Wang等[10]通過TPR和ESR等手段證明,V4+能穩定存在于VOx/Al2O3催化劑的表面。Fu等[11]發現,SnO2摻雜能提高氧化釩在氧化鋁表面的分散狀態,同時與釩物種的相互作用提高了催化劑的還原性,實現了催化性能的改善。由此可見,負載型釩基催化劑的氧化釩分散狀態、表面酸性和釩價態直接關系到催化劑的活性和穩定性。
本工作以γ-Al2O3為載體,采用等體積浸漬法制備了負載高分散氧化釩催化劑(V2O5/γ-Al2O3),并通過K,Ca,Mg對V2O5/γ-Al2O3的表面酸性及釩價態進行調控,研究催化劑表面酸性改性的變化規律及與異丁烷脫氫活性和選擇性之間的構效關系。采用XRD、XPS、N2吸附-脫附、NH3-TPD和H2-TPR等方法對催化劑進行表征。
1 實驗部分
1.1 試劑及儀器
偏釩酸銨:AR,阿拉丁化學試劑上海有限公司;草酸:AR,上海強順化學試劑有限公司;硝酸鉀:AR,國藥集團化學試劑有限公司;γ-Al2O3載體:國藥集團化學試劑有限公司。
采用Bruker公司AXS D8 Advance型X射線衍射儀進行催化劑的XRD分析,CuKα射線,管電壓40 kV,管電流40 mA,PSD檢測;采用Perkin-Elmer公司PHI 5000 ESCASystem型X射線光電子能譜儀進行催化劑的XPS分析,AlKα射線,通能為46.95 eV;采用Micromeritics公司Tri Star 3000型自動物理吸附儀進行催化劑的N2吸附-脫附測定,測量前將催化劑在真空、160 ℃下預處理2 h,再于液氮溫度下,用高純氮進行吸附和脫附測定;采用Micromeritics公司Auto Chem 2920型氨程序升溫脫附裝置進行NH3-TPD測試,試樣在高純He氣氛、550 ℃下吹掃2 h,冷卻至室溫,通入過量NH3直至吸附飽和,繼續吹掃2 h以除去物理吸附的NH3,脫附過程由室溫以10 ℃/min的速率升至500 ℃;采用Micromeritics公司ChemSorb2720型化學吸附儀進行H2-TPR測定,將約0.1 g試樣裝入U形石英管內,He氣氛、600 ℃下預處理1 h,冷卻至20 ℃,切換至H2/Ar混合氣后,以10 ℃/min的升溫速率在20~980 ℃區間記錄H2還原信號;采用Thermo Fisher公司Thermo Trace Ultra GC型氣相色譜儀對產物氣體實現在線全分析,經氣動六通閥自動采樣,FID檢測,Varian Plot Q色譜柱(50 m×0.32 mm×10 μm)。
1.2 催化劑的制備
稱取一定量的偏釩酸銨和草酸于燒杯中,偏釩酸銨和草酸的摩爾比為1:2。加入一定量的蒸餾水(其量約為γ-Al2O3飽和吸附量的1.2倍),在80 ℃下攪拌至草酸和偏釩酸銨全部溶解。在溶液中加入額定量的γ-Al2O3載體,在緩慢攪拌中實現均勻浸漬,然后在靜置狀態下浸漬12 h。浸漬后載體于120 ℃下烘干,再于550 ℃下焙燒4 h。以該方法制得的催化劑標記為a%VOx/γ-Al2O3,其中,a為3,5,7,9,11,分別表示釩占催化劑的質量分數。
以7%VOx/γ-Al2O3催化劑為母體,以硝酸鉀為前體,采用等體積浸漬方法對γ-Al2O3負載氧化釩催化劑進行改性。稱取一定量的硝酸鉀,量取與母體催化劑等體積的去離子水在室溫下進行溶解,然后將溶液緩慢滴加于7%VOx/γ-Al2O3催化劑中。浸漬鉀后的催化劑于120 ℃下烘干,然后在空氣氣氛、550 ℃下焙燒4 h。以該方法制得的催化劑標記為b%K-7%VOx/γ-Al2O3,其中,b=0.5,1.0,1.5,2.0,分別表示K占催化劑的質量分數。采用同樣方法制備的Ca和Mg改性催化劑分別標記為1.5%Ca-7%VOx/γ-Al2O3和1.5%Mg-7%VOx/γ-Al2O3。
1.3 催化劑性能的評價
將上述催化劑應用于異丁烷催化脫氫制備異丁烯反應,采用全自動連續流動固定床反應裝置評價催化劑性能。反應在常壓下進行,反應管內徑為8 mm,催化劑裝填量為2.0 g。先在5%(φ)H2和95%(φ)N2氣氛下,以10 ℃/min的速率將反應爐溫度升到550 ℃,然后N2和原料氣i-C4H10以15:1摩爾比混合進入反應管,反應床層溫度由反應系統自動控制。
2 結果與討論
2.1 V2O5在γ-Al2O3上的分散及其催化性能
氧化釩的表面分散狀態對于脫氫催化劑的活性和穩定性有著重要的影響[12]。劉堅等[13]發現,以VOx/SBA-15為丙烷氧化脫氫催化劑,低釩負載量(w)(x<0.1%)可帶來更高的醛類選擇性,而中等的釩負載量則帶來更高的丙烷脫氫深度氧化活性,高含量的釩負載量會抑制反應的活性。氧化釩同載體之間的相互作用也會影響氧化釩在催化劑表面的分散狀態,帶來脫氫活性的變化。由于氧化釩的塔曼溫度低于400 ℃,在焙燒時會在載體表面發生自擴散現象,由于載體表面性質不同,氧化釩會產生不同的分散特性。Bond等[12]發現,當釩的負載量處在一個合適的范圍時,釩能夠在載體上呈最大程度的單層分散,而超過了這一界限,釩物種在表面會發生堆積覆蓋,進而形成氧化釩結晶,導致催化劑活性降低。H?j等[14]通過噴霧熱解法研究發現,隨著負載量的升高,氧化釩由分散分布過渡到低聚分散,最終在負載量超過10%后出現晶粒。王玨等[15]采用XRD方法對金屬氧化物在HZSM-5表面的MgO單層分散閾值進行了研究。本工作應用類似的方法將不同負載量的氧化釩負載在氧化鋁載體上,然后應用XRD對載體表面的氧化釩的分散狀態進行了表征。
圖1為負載量3%~9%的a%VOx/γ-Al2O3系列催化劑的XRD譜圖。由圖1可知,在負載量低于7%時,XRD圖譜中只出現了γ-Al2O3的衍射信號,未觀察到氧化釩物種的衍射峰,這表明氧化釩在載體表面處于良好的分散狀態;當釩負載量到達9%時,氧化釩物種的衍射峰開始出現。這表明氧化釩物種已經在載體表面發生多層負載,導致氧化釩晶體的聚集。因此,當氧化釩負載量低于7%時,氧化釩在γ-Al2O3表面處于良好的分散狀態。
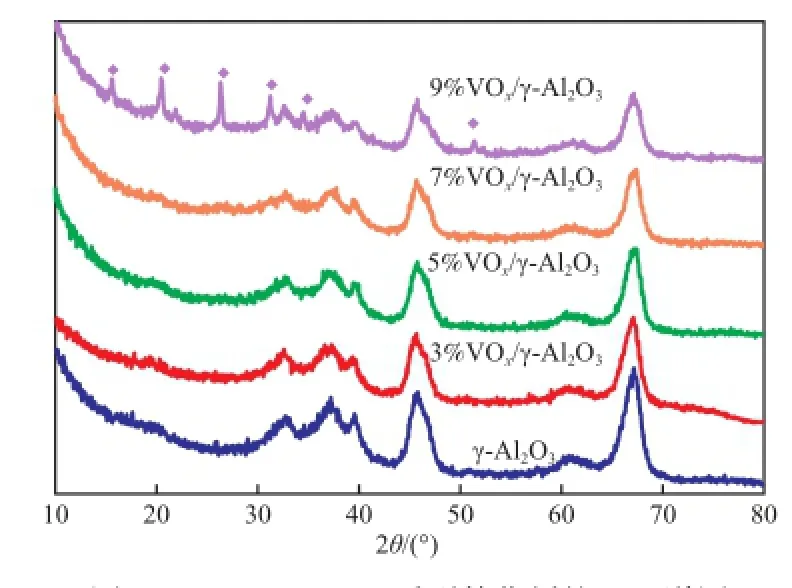
圖1 a%VOx/γ-Al2O3系列催化劑的XRD譜圖Fig.1 XRD patterns ofa%VOx/γ-Al2O3catalysts. a:the mass content of vandium in the catalyst.◆ V2O5
表1為a%V2O5/γ-Al2O3系列催化劑的比表面積和反應活性數據。由表1可見,隨著釩負載量的升高,催化劑比表面積逐漸降低。當釩負載量在0~7%之間時,比表面積下降幅度較大,比表面積由210 m2/g降至170 m2/g,當釩負載量從7%增至9%時,比表面從170 m2/g略微下降至166 m2/g。該結果同樣反映出氧化釩在γ-Al2O3表面的分散狀態發生了顯著改變。氧化釩在載體表面的分散狀態直接關系到催化劑的反應性能,由表1給出的活性結果可見,氧化鋁對于異丁烷脫氫沒有活性,氧化釩負載量的變化對異丁烷脫氫的反應活性影響顯著。7%VOx/γ-Al2O3催化劑具有最高的轉化率和異丁烯選擇性,分別為36.2%和88.1%。當氧化釩的負載量從3%提高至7%時,異丁烷的轉化率逐漸升高,異丁烯選擇性相對穩定。當氧化釩負載量為9%時,異丁烷的轉化率降至23.1%,異丁烯選擇性也出現明顯的下降趨勢。因此,異丁烷的催化脫氫活性與氧化釩在載體表面的分散狀態密切相關,當氧化釩在氧化鋁表面處于良好分散狀態(7%VOx/γ-Al2O3)時,催化劑保持最佳的反應活性和選擇性。
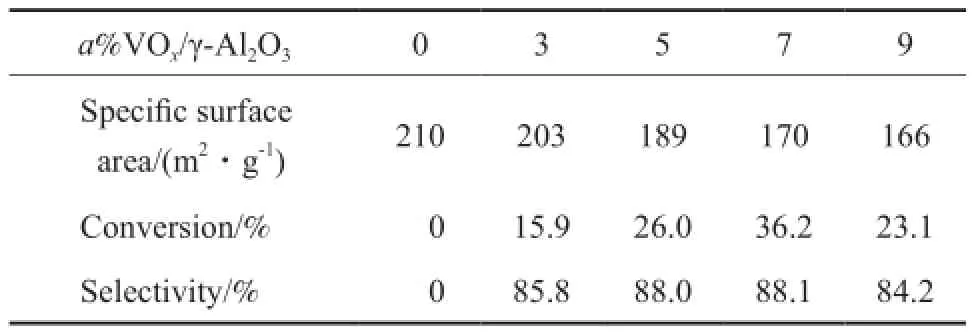
表1a%VOx/γ-Al2O3系列催化劑的比表面積和活性數據Table 1 Specifc surface area and activity of thea%V2O5/Al2O3catalysts
2.2 催化劑表征及其性能
2.2.1 K,Ca,Mg對催化劑表面酸性的調變作用
釩氧化物種在載體表面的分散狀態和結構形式主要取決于載體的性質和負載量。Blasco等[16]研究了釩氧化物種在不同酸堿載體上的結構和分散狀態,認為釩氧化物種在酸性載體表面易形成V2O5晶相,在堿性載體上易形成金屬釩酸鹽。Cortright等[17]研究發現,利用K2O對Pt-Sn/Al2O3催化劑改性可同時提高異丁烷的脫氫活性和選擇性,其主要原因是通過堿金屬元素改性一定程度上減少了酸性載體上活性金屬的團聚,增加了催化劑活性表面。因此,釩氧化物種在載體表面的分散狀態和表面酸性是決定異丁烷脫氫活性和穩定性的關鍵。本工作以γ-Al2O3負載高分散氧化釩為母體催化劑,通過等體積浸漬法對催化劑表面酸性進行調控,研究堿金屬K和堿土金屬Ca和Mg對催化劑表面酸性改性的變化規律。
采用NH3-TPD測定K對7%VOx/γ-Al2O3系列催化劑改性前后的NH3脫附溫度和脫附量,結果見表2。由表2可知,在NH3脫附過程中,在160~171 ℃和630~645 ℃溫區出現2個脫附峰,分別代表催化劑表面弱酸和強酸中心。可見,K的添加對催化劑表面酸量的抑制特別明顯,當K添加量為1.0%時,強酸中心就幾乎被全部中和,弱酸量僅為改性前的50%。經2.0%的K修飾后的7%VOx/γ-Al2O3表面總酸量從0.725 mmol/g下降至0.291 mmol/g。與此同時,NH3的脫附溫度隨著K添加量的增加向低溫方向偏移,表明催化劑表面酸強度在減弱。由表2還可知,K改性使得催化劑的比表面積略有提高,但隨著K添加量的提高比表面積呈下降趨勢。當K添加量大于2.0%時,下降趨勢顯著增加。為了更好地研究負載型氧化釩表面酸性對反應性能的影響,分別制備了1.5%Ca和Mg改性的7%VOx/γ-Al2O3催化劑,并對催化劑表面酸性進行了對比研究。實驗結果表明,相對于同樣負載量的K改性,1.5%的Ca和1.5%的Mg改性的7%VOx/γ-Al2O3對催化劑表面酸性的調變作用較弱,改性后催化劑表面的強酸中心和弱酸中心有所下降,總酸量的下降幅度分別為22.8%和9.5%。NH3脫附峰溫度變化不大,這表明堿金屬和堿土金屬元素都可實現對7%VOx/γ-Al2O3系列催化劑表面酸性的調變,對于催化劑表面酸量和酸強度的調節作用強弱次序為:K > Ca > Mg。

表2 催化劑的NH3-TPD表征結果Table 2 NH3-TPD results of some catalysts
2.2.2 K,Ca,Mg對催化劑中釩價態的調節作用
Wachs[18]通過拉曼、紅外光譜等手段發現,K的加入能夠改變V=O鍵的鍵合強度,這說明K元素不僅起到改變催化劑表面酸堿性的作用,在調節催化劑表面分散狀態、電子性能等方面也起到一定作用。Harlin等[19]研究了釩氧化鋁催化劑上V價態對于異丁烷脫氫的影響,研究結果表明:氧化釩在充分焙燒后主要以V5+存在,在異丁烷脫氫的過程中會被還原成更低的價態,催化劑的活性和V的還原價態有關,V的活性價態在3+和4+之間,在催化劑中添加Mg和Zr等金屬能影響還原后V的價態,從而影響催化劑的轉化率和選擇性。
為了研究堿金屬元素對V2O5/γ-Al2O3中V價態的調變作用,將不同添加量的K添加到7%VOx/ γ-Al2O3催化劑中,通過H2-TPR實驗計算釩氧化態的變化。圖2(A)是K改性前后7%VOx/γ-Al2O3催化劑的H2-TPR表征結果。由圖2(A)可知,K的添加使得7%VOx/γ-Al2O3中氧化釩的還原程度顯著降低,且隨著K添加量的增加,還原峰面積逐步降低。從還原峰溫度來看,催化劑的還原溫度隨K添加量的增加逐漸提高。結合表3中數據可知,當K的添加量為2.0%時,氧化釩的還原程度被嚴重抑制,其還原量僅為7%VOx/γ-Al2O3的50%。K改性導致催化劑還原量的降低說明催化劑表面V的氧化價態降低程度減小,氧化釩還原峰溫的提高表明了氧化釩和載體之間的相互作用力得到增強。圖2(B)比較了添加量為1.5%的K,Ca,Mg對7%VOx/ γ-Al2O3催化劑改性前后的H2-TPR表征結果。由圖2(B)可知,同等添加量的K,Ca,Mg對7%VOx/ γ-Al2O3催化劑的改性均導致還原程度的降低,其中,Ca和Mg改性的催化劑H2還原量約為0.8 mmol/ g,略大于K改性的催化劑H2還原量(0.7 mmol/g),還原峰溫的高低次序為:K>Ca>Mg。可見,堿金屬K對7%VOx/γ-Al2O3催化劑的改性作用強于Ca和Mg。
根據Harlin等[19]報道的方法,將H2-TPR實驗中的H2消耗量和V的價態進行關聯,假設每摩爾的V5+價態降低至V4+價態將消耗0.5 mol的H2來計算還原后V的價態。表3為經不同K添加量改性后7%VOx/γ-Al2O3催化劑的H2還原量和還原后V的平均價態。由表3可知,未經改性的7%VOx/γ-Al2O3催化劑還原后,其V的價態為2.87+;經K改性后V的還原價態明顯增加,當K的添加量在0.5%~1.5%之間時,V的平均價態在2.96+~3.95+價之間。同時還原峰溫向高溫方向移動,這表明了氧化釩和載體結合力增強。當K的添加量為2.0%時,V的平均價態為4.64+,還原峰溫達到最大值545 ℃,表明當K的添加量較高時,也可能發生K與氧化釩在載體表面形成釩酸鹽[16],導致了氧化釩還原程度的顯著降低。由表3還可知添加量為1.5%的Ca和Mg改性的7%V2O5/γ-Al2O3催化劑還原后V的價態。添加量為1.5%的K改性后V的還原價態為3.95+,同樣添加量的Ca和Mg改性后的V的還原價態為3.82+和3.83+。與K相比,Ca和Mg對V還原后價態的調節作用相對較弱。
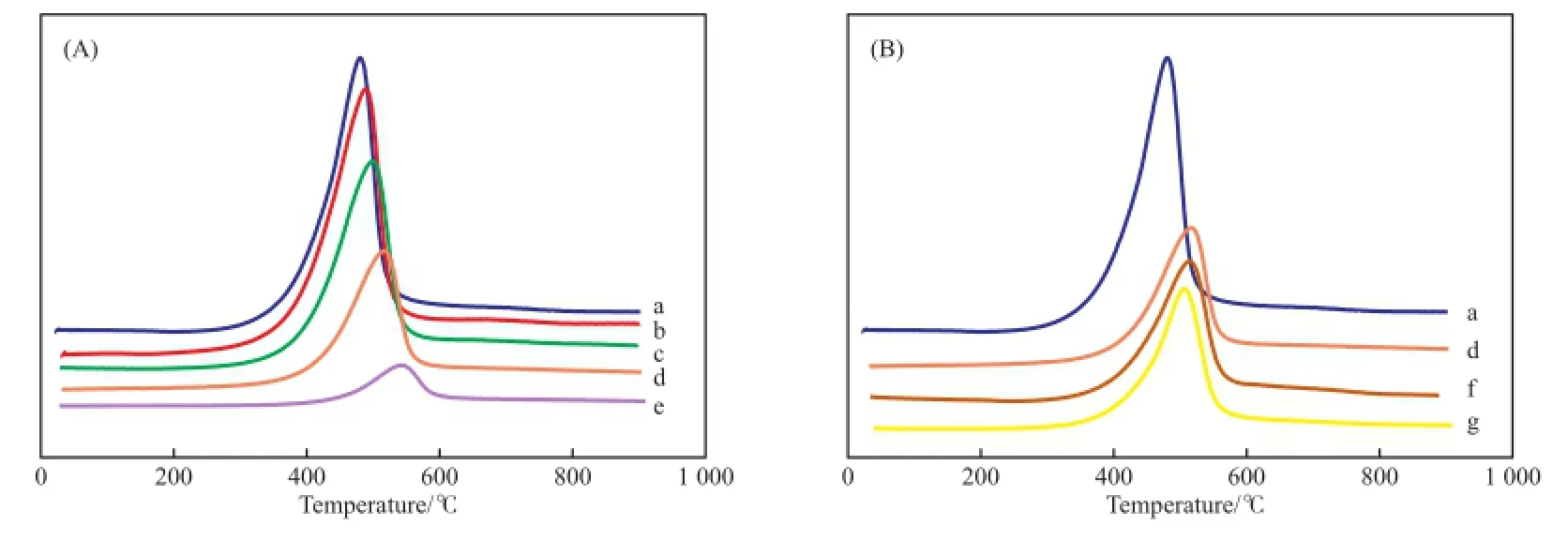
圖2 催化劑的H2-TPR表征結果Fig.2 H2-TPR results of the catalysts.a 7%VOx/γ-Al2O3;b 0.5%K-7%VOx/γ-Al2O3;c 1.0%K-7%VOx/γ-Al2O3;d 1.5%K-7%VOx/γ-Al2O3;e 2.0%K-7%VOx/γ-Al2O3;f 1.5%Ca-7% VOx/γ-Al2O3;g 1.5%Mg-7%VOx/γ-Al2O3
綜上,堿金屬和堿土金屬對7%VOx/γ-Al2O3催化劑的改性作用不僅是對表面酸性的調控,還增強了氧化釩與載體的相互作用,提高了還原態V的價態,使V的價態處于具有更好催化性能的3+和4+之間。

表3 TPR法計算催化劑中V的平均氧化態Table 3 Average oxidation state of vanadium in the catalysts calculated by TPR
2.3 催化劑活性和穩定性
催化劑的異丁烷脫氫反應活性見表4。表4顯示出K,Ca,Mg改性前后7%VOx/γ-Al2O3催化劑對異丁烷脫氫的活性評價結果。由表4可知,K改性對于催化劑的反應初活性有一定程度的抑制作用,當K的添加量為0.5%~1.5%時,異丁烷的轉化率下降了約10百分點,但催化劑對于異丁烷轉化的選擇性顯著提高,異丁烯選擇性從原來的88%上升到97%左右。值得關注的是經190 min反應后,K-7%VOx/γ-Al2O3催化劑活性仍然保持穩定,異丁烷的轉化率保持上升的趨勢,高于異丁烷在7%VOx/γ-Al2O3催化劑上的轉化率。隨著反應的進行,異丁烯的選擇性表現出不同的特征,在0.5%~1.0%K改性的7%VOx/γ-Al2O3催化劑上,經190 min反應后異丁烯的選擇性有所下降,而經1.5%K改性的7%VOx/γ-Al2O3催化劑,異丁烯的選擇性仍然保持在97%以上。但當K的添加量為2.0%時,轉化率從改性前的36.2%下降到14.9%。造成這一現象的原因和催化劑表面酸中心的減少或是釩酸鹽的存在密切相關。在同樣反應條件下比較了1.5%Ca-7%VOx/γ-Al2O3和1.5%Mg-7%VOx/γ-Al2O3催化劑對異丁烷脫氫性能。研究結果表明,與K相比,負載量相同時Ca和Mg的改性作用略弱,反應初活性影響較小,選擇性的增幅較小。負載量為1.5% 時,Ca和Mg改性催化劑異丁烷10 min轉化率分別為30.1%和34.9%,高于K改性催化劑。經190 min反應后轉化率分別保持在30.7%和28.7%。負載量為1.5% 時,Ca和Mg改性對催化劑的異丁烯選擇性具有顯著的提升作用,都保持在95%以上但略低于K改性催化劑的97.9%選擇性。負載量相同時,Ca和Mg對7%VOx/γ-Al2O3催化劑的改性作用比K弱,這與催化劑的表征結果相一致。

表4 催化劑的異丁烷脫氫反應活性Table 4 Activities of the catalysts for the dehydrogenation of isobutane
圖3為1.5%K-7%VOx/γ-Al2O3催化劑活性穩定性的評價結果。
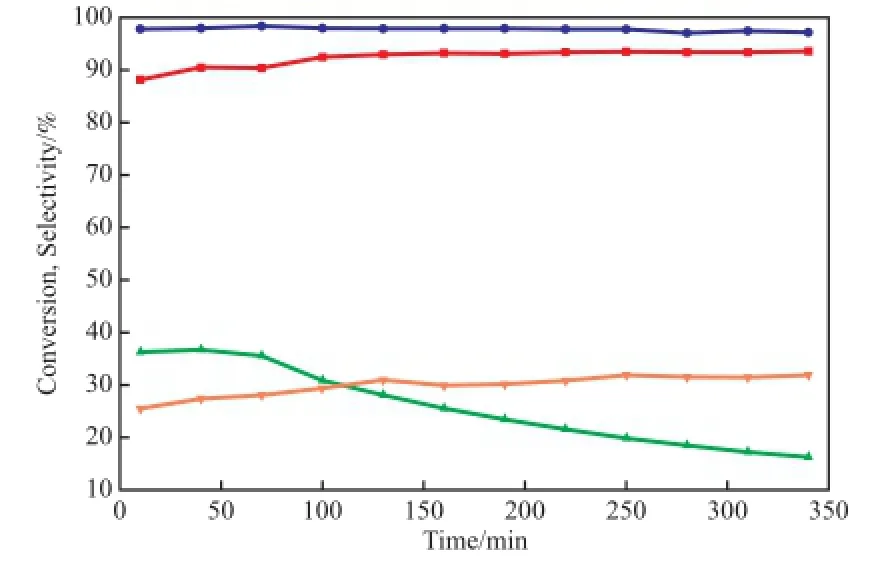
圖3 催化劑穩定性的評價結果Fig.3 Stability of the catalysts.● Selectivity to isobutene on 1.5%K-7%VOx/γ-Al2O3;■ Selectivity to isobutene on 7%VOx/γ-Al2O3;▼ Conversion of isobutane on 1.5%K-7%VOx/γ-Al2O3;▲ Conversion of isobutane on 7%VOx/γ-Al2O3
由圖3可知,經負載量為1.5%的K改性的7%VOx/γ-Al2O3催化劑雖然初活性比7%VOx/ γ-Al2O3催化劑活性低,但在340 min反應范圍內保持了良好的穩定性能,反應的轉化率保持在30%以上。而7%VOx/γ-Al2O3催化劑的活性持續下降,在反應110 min后其轉化率已經低于1.5%K-7% VOx/ γ-Al2O3催化劑,經340 min后反應轉化率下降至16%。1.5%K-7%VOx/γ-Al2O3催化劑對于異丁烯的選擇性普遍高于改性前催化劑,異丁烯的選擇性保持在97%以上。
K,Ca,Mg改性的V2O5/γ-Al2O3催化劑的活性與催化劑表面酸性和V價態密切相關。馬紅超等[4]采用ESR技術對負載性V2O5/γ-Al2O3催化劑焙燒和還原后V的超精細結構進行了研究。研究結果表明,V4+超精細結構與活性物種在γ-Al2O3表面的分散有對應關系。Harlin等[6]應用XPS技術對V2O5/γ-Al2O3的還原價態進行了深入研究,指出焙燒后的氧化釩主要以V5+的形式存在,經H2還原后價態主要在V4+~V3+之間,異丁烷的脫氫活性與氧化釩的還原價態有密切關系。
綜上, K對V2O5/γ-Al2O3催化劑表面的酸量和酸強度有顯著的調節作用,極大降低了催化劑表面的強酸性位,這可有效抑制副反應和積碳,使催化劑具有更好的選擇性和穩定性,但抑制副反應的同時也會使催化劑的初始轉化率降低。H2-TPR表征結果顯示,當K的添加量為1.5%時,催化劑表面的強酸位基本被中和,但仍具有相當量的弱酸位,同時V的還原價態為3.95+,這使得催化劑具有最佳的反應活性和選擇性。Ca和Mg同樣可對催化劑的表面酸性和釩還原價態進行調控,但與K相比,其調控作用略弱。
3 結論
1) 異丁烷的催化脫氫活性與氧化釩在載體表面的分散狀態密切相關,當氧化釩負載量低于7%且氧化釩在氧化鋁表面處于良好分散狀態時,催化劑保持最佳的反應活性和選擇性。
2) K對V2O5/γ-Al2O3催化劑表面的酸性和V的還原價態具有顯著的調節作用,表面強酸位的減少和V還原價態的提高,可有效地提高催化劑的選擇性和穩定性;當K添加量為1.5%時,強酸中心幾乎被全部抑制,同時保留了適量的弱酸中心,而V的還原價態由2.87+調變為3.95+,這使催化劑的性能得到顯著提高;在1.5%K-V2O5/γ-Al2O3催化劑上異丁烷脫氫反應340 min后,異丁烷轉化率保持在30%以上,異丁烯選擇性保持在97%以上。
3) Ca和Mg同樣可以對V2O5/γ-Al2O3催化劑的表面酸性和釩還原價態進行調控,但與K相比,其調控作用略弱。
[1]Cortright R D,Dumesic J A. Microcalorimetric,spectroscopy,and kinetic-studies of silica-supported PT and PT/SN catalysts for isobutane dehydrogenation[J]. J Catal,1994,148(2):771 - 778.
[2]Cortright R D,Levin P E,Dumesic J A. Kinetic studies of isobutane dehydrogenation and isobutene hydrogenation over Pt/Sn-based catalysts[J]. Ind Eng Chem Res,1998,27(5):1717 - 1723.
[3]Bhasin M,McCain J,Vora B,et al. Dehydrogention and oxydehydrogenation of parrifns to olefns[J]. Appl Catal,A, 2001,221(1):397 - 419.
[4]Nawaz Z. Light alkane dehydrogenation to light olefn technologies:A comprehensive review[J]. Rev Chem Eng,2015,31(5):413 - 436.
[5]Carrero C A,Schloegl R,Wachs I E,et al. Critical literature review of the kinetics for the oxidative dehydrogenation of propane over well-defned supported vanadium oxide catalysts[J]. ACS Catal,2014,4(10):3357 - 3380.
[6]Ogonowski J,Skrzynska E. Dehydrogenationof isobutane in the presence of carbon dioxide over supported vanadium oxide catalysts[J]. React Kinet Catal Lett,2006,88(2):293 - 300.
[7]Vora B V. Development of dehydrogenation catalysis and processes[J]. Top Catal,2012,55(19):1297 - 1308.
[8]Volpe M,Tonetto G,de Lasa H. Butane dehydrogenation on vanadium supported catalysts under oxygen free atmosphere[J]. Appl Catal,A,2004,272(1/2):69 - 78.
[9]Harlin M E,Niemi V M,Krause A O I. Alumina-supported vanadium oxide in the dehydrogenation of butanes[J]. J Catal,2000,195(1):67 - 68.
[10]Wang Guojia,Ma Hongchao,Li Ying,et al. Dehydrogenation of isobutane over V2O5/gamma-Al2O3catalyst[J]. React Kinet Catal Lett,2001,74(1):103 - 110.
[11]Fu Yinghuan,Ma Hongchao,Wang Zhenlu,et al. Charaterization and reactivity of SnO2-doped V2O5/gamma-Al2O3catalysts in dehydrogenation of isobutane to isobutene[J]. J Mol Catal,A:Chem,2004,221(1/2):163 - 168.
[12]Bond G C,Tahir S F. Vanadium-oxide monolyer catalysts-preparation,characterization and catalytic activity[J]. Appl Catal,1991,71(1):1 - 31.
[13]劉堅,趙震,王宏宣,等. 擔載釩氧化物催化劑對丙烷選擇氧化性能[J]. 物理化學學報,2011,27(11):2659 - 2664.
[14]H?j M,Jensen A D,Grunwaldt J. Structure of alumina supported vanadium catalysts for oxidative dehydrogenation of propane prepared by fame spray pyrolysis[J]. Appl Catal,A,2013,451,207 - 215.
[15]王玨,趙碧英,謝有暢. MgO/HZSM-5中MgO分散狀態和催化性能的關系[J]. 物理化學學報,2001,17(11),966 -971.
[16]Blasco T,Lopez N J M. Oxidative dehydrogenation of short chain alkane on supported vanadium oxide catalysts[J]. Appl Catal,A,1997,157(1/2):117 - 142.
[17]Cortright R D,Dumesic J A. Efect of potassium on silica-supported Pt and Pt/Sn catalysts for isobutane dehydrogenation[J]. J Catal,1995,157(2):576 - 583.
[18]Wachs I E. Recent conceptual advances in the catalysis science of mixed oxide catalytic materials[J]. Catal Today,2005,100(1/2):79 - 94.
[19]Harlin M,Niemi V,Krause A,et al. Effect of Mg and Zr modifcation on the activity of VOx/Al2O3catalysts in the dehydrogenation of butanes[J]. J Catal,2001,203(1):242 -252.
(編輯 楊天予)
Modification of acidity and vanadium valence of V2O5/γ-Al2O3catalyst for dehydrogenation of isobutane
Zhang Lin,Qin Feng,Huang Zhen,Shen Wei,Xu Hualong
(The Department of Chemistry,Fudan University,Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials,Laboratory of Advanced Materials of Fudan University,Shanghai 200433,China)
Highly dispersed V2O5/γ-Al2O3catalysts for the dehydrogenation of isobutane were prepared through impregnation,and K,Ca and Mg were introduced to modify the surface acidity and vanadium valence of the V2O5/γ-Al2O3catalysts. The catalysts were characterized by means of XRD,XPS,N2adsorption and desorption, NH3-TPD,and H2-TPR. It was indicated that,when the vanadium loading was less than 7%(w),the vanadium oxide dispersed well on the γ-Al2O3support and the catalysts showed high activity in the dehydrogenation of isobutane to isobutene. The characterization results revealed that the potassium modification had significant adjustment ef ects on the surface acidity and vanadium valence of the catalysts. The strong acid sites were nearly neutralized when the loading of potassium was 1.5%(w),while the moderate amount of weak acid sites was kept on the catalyst surface and the vanadium valence was adjusted from 2.87+ to 3.95+. After the dehydrogenation over the 1.5%(w)K-V2O5/γ-Al2O3catalyst was conducted for 340 minutes,the conversion of isobutane and the selectivity to isobutene were still kept above 30% and more than 97%,respectively.
V2O5/γ-Al2O3catalyst;surface acidity;vanadium valence;isobutane dehydrogenation
1000 - 8144(2016)09 - 1043 - 07
TQ 223.12
A
10.3969/j.issn.1000-8144.2016.09.004
2016 - 03 - 17;[修改稿日期]2016 - 06 - 15。
張林(1988—),男,山東省煙臺市人,碩士生,電話 021 - 65642401,電郵 11210220070@fudan.edu.cn。聯系人:徐華龍,電話 021 - 65642401,電郵 shuhl@fudan.edu.cn。
上海市科學技術委員會資助國際合作項目(14120700700);重點實驗室基金項目(14DZ2273900)。
猜你喜歡
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
中國塑料(2016年12期)2016-06-15 20:30:07
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
中國塑料(2016年5期)2016-04-16 05:25:36
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
中國塑料(2015年3期)2015-11-27 03:41:38
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
