健脾顆粒的質(zhì)量標(biāo)準(zhǔn)研究
2017-02-21 19:12:00李佳杏孫蓉??
中國民族民間醫(yī)藥·上半月 2017年1期
李佳杏++孫蓉??

【摘要】目的:建立控制健脾顆粒的質(zhì)量標(biāo)準(zhǔn)。方法:采用薄層色譜法對健脾顆粒中白術(shù)、橙皮苷進行鑒別;用高效液相色譜法測定制劑中橙皮苷的含量,采用十八烷基硅烷鍵合硅膠柱(Agilent XDB C18柱;4.6mm×250mm,5μm),流動相為乙腈-0.2%磷酸溶液(19∶[KG-*3/5]81),流速為1.0mL/min,柱溫為30℃,檢測波長:283nm。結(jié)果:定性鑒別方法能檢出白術(shù)、橙皮苷,且薄層色譜中陰性無干擾,專屬性強;橙皮苷的線性范圍為0.131~4.192mg,平均加樣回收率為100.26%,RSD為2.21%。結(jié)論:該方法簡便、準(zhǔn)確,重現(xiàn)性好,可用作健脾顆粒的質(zhì)量控制。
【關(guān)鍵詞】健脾顆粒;橙皮苷;白術(shù); HPLC
【中圖分類號】R914.1【文獻標(biāo)志碼】 A【文章編號】1007-8517(2017)01-0023-03
Abstract:
Keywords:
健脾顆粒的處方來源于部頒中藥成方制劑第二冊[1](標(biāo)準(zhǔn)號:WS3-B-0381-90),由黨參、炒白術(shù)、陳皮等6味藥組成,具有健脾開胃的功效,臨床用于脾胃虛弱,脘腹脹滿,食少便溏。原標(biāo)準(zhǔn)中無含量測定項和薄層色譜鑒別項,為了能全面地控制其藥品質(zhì)量,本文通過對處方中的組分進行薄層色譜鑒別試驗研究,確定白術(shù)、橙皮苷的薄層色譜鑒別方法。同時對陳皮、枳實中橙皮苷進行了含量測定試驗。通過多次反復(fù)試驗,確定該方法簡捷,效果好,可用于本品的質(zhì)量控制方法。
1儀器與材料
1.1儀器B3200S-T超聲機(上海必能信超聲);高效液相色譜儀(美國Agilent 1100液相色譜儀系統(tǒng))。
1.2材料橙皮苷對照品(批號:110712-201010)、白術(shù)對照藥材(批號:120925-201109)等均購自中國藥品生物制品檢定所;健脾顆粒(昆明中藥廠有限公司提供,規(guī)格:5g/袋);高效進樣使用的乙腈、甲醇為色譜純;其他試劑均為分析純。
2方法與結(jié)果
2.1薄層色譜鑒別
2.1.1白術(shù)的薄層色譜鑒別取本品10g,研細,加40mL乙酸乙酯和5mL水,振搖2min,傾取上清液,殘渣續(xù)加乙酸乙酯30mL,振搖1min,合并上清液,每次分別用20mL 3%碳酸鈉溶液振搖提取3次,取堿水液,用濃鹽酸調(diào)pH值至2~3,每次用20mL乙酸乙酯振搖提取3次,分取乙酸乙酯液,用水30mL洗滌,分取乙酸乙酯液,蒸至近干,加甲醇0.5mL制成供試品溶液[2]。另外取0.5g白術(shù)對照藥材,加30mL水置150mL燒杯中煮沸,保持微沸30 min,濾過后保留殘渣繼續(xù)加水20mL煮沸,保持微沸20min,用脫脂棉濾過煎液,合并剩余的煎液并濃縮至約1mL,放冷,加乙酸乙酯15mL混合,“自振搖2min”起,同法制成對照藥材溶液。照薄層色譜法(《中國藥典》2015年版四部通則0502)試驗[3],吸取上述供試品溶液15~20μL,白術(shù)對照藥材溶液10μl,分別點于同一硅膠G薄層板上,以環(huán)己烷-乙酸乙酯(7∶3)為展開劑,展開,取出,晾干。噴以10%硫酸乙醇溶液,于105℃加熱3~5 min,置紫外光燈(365nm)下檢視。供試品色譜中,在與對照藥材色譜相應(yīng)的位置上,顯相同顏色的熒光斑點。結(jié)果見圖1。
2.1.2橙皮苷的鑒別取適量本品研細,稱取2g,加20mL甲醇,超聲30min,用定性濾紙濾過,將濾液蒸至近干,加2mL甲醇溶解制成供試品溶液。另取橙皮苷對照品適量,加甲醇制成飽和溶液,作為對照品溶液[2]。照薄層色譜法(《中國藥典》2015年版四部通則0502)試驗[3],吸取供試品溶液2~4μL,對照品溶液4μL,分別點于同一硅膠G薄層板上,以乙酸乙酯-甲醇-水(100∶[KG-*3/5]17∶[KG-*3/5]13)為展開劑,展開至約3cm,取出,晾干,再以甲苯-乙酸乙酯-甲酸-水(20∶[KG-*3/5]10∶[KG-*3/5]1∶[KG-*3/5]1)的上層溶液為展開劑,展至8cm,取出,晾干,噴以三氯化鋁試液,置紫外光燈(365nm)下檢視。供試品色譜中,在與對照品色譜相應(yīng)的位置上,顯相同顏色的熒光斑點。結(jié)果見圖2。
2.2含量測定
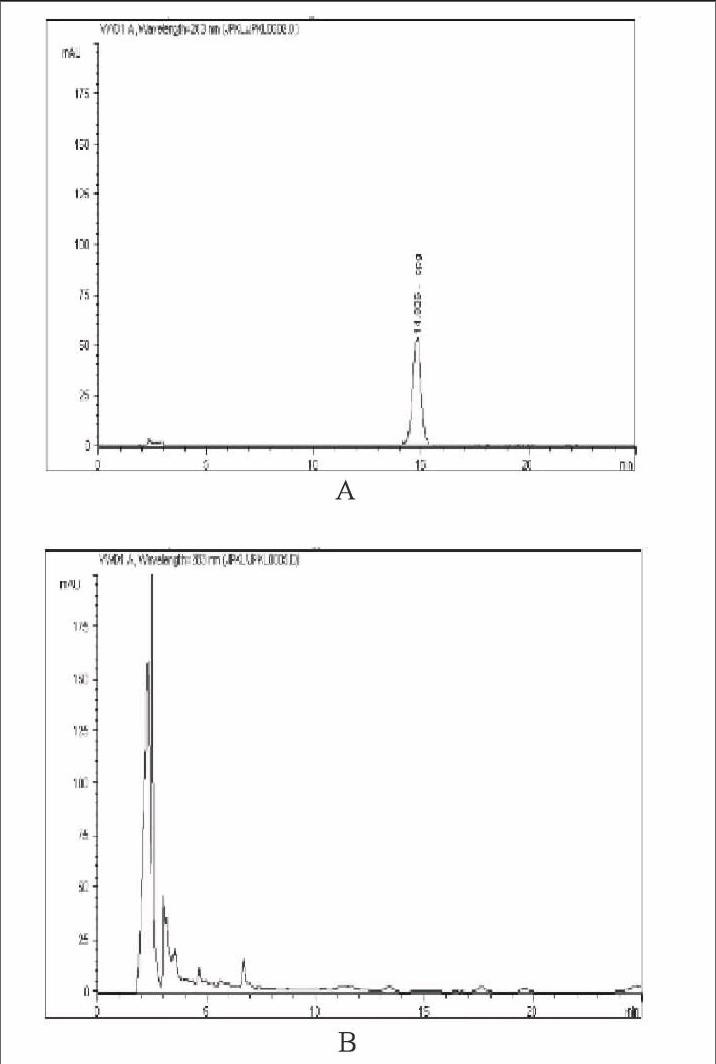
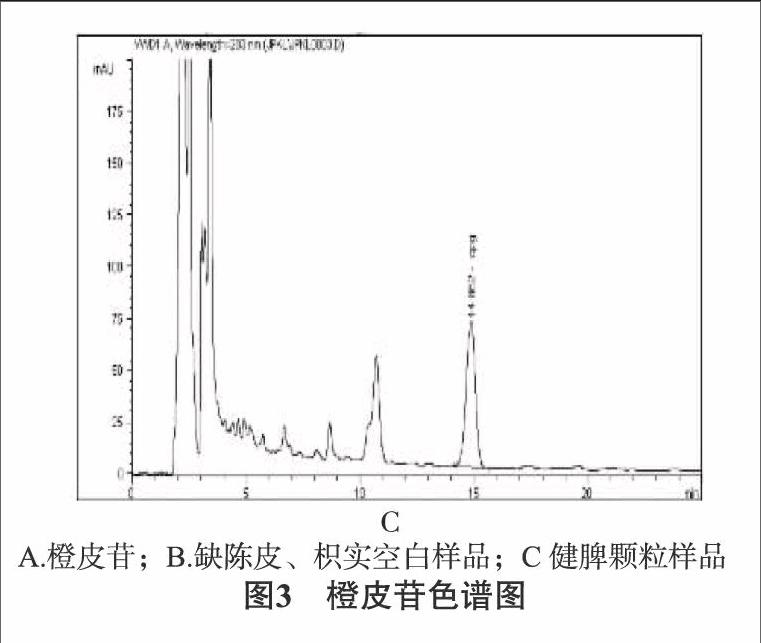
2.2.1色譜條件色譜柱:采用十八烷基硅烷鍵合硅膠柱(Agilent ZORBAX Eclipse XDB-C18柱),流動相為乙腈-0.2%磷酸溶液(19∶[KG-*3/5]81),流速為1.0mL/min,柱溫為30℃,檢測波長:283nm。按橙皮苷峰的理論塔板數(shù)計算應(yīng)不應(yīng)低于2000。見圖3。
2.2.2對照品溶液的制備精密稱取橙皮苷對照品適量,加甲醇溶解,制成每1mL含524μg的溶液,即得[2]。
2.2.3供試品溶液的制備取本品適量,研細,取2g,精密稱定,置具塞錐形瓶中,精密加入70%乙醇25mL,密塞,稱定重量,超聲處理(功率160W,頻率50kHz)30min,放冷,再稱定重量,用70%乙醇補足減失的重量,搖勻,靜置,取上清液,濾過,取續(xù)濾液,即得。
2.2.4陰性對照溶液的制備取處方中除陳皮、枳實外的其他藥材,按處方劑量及制法和工藝要求制備缺陳皮、枳實的空白樣品,照含量測定項下供試品溶液的制備方法制成陰性對照樣品溶液,同法進行測定,結(jié)果在與對照品橙皮苷相同的保留時間位置,無其他干擾峰出現(xiàn),表明除陳皮、枳實外的其他藥材和輔料對橙皮苷測定無干擾。見圖3。
2.2.5線性關(guān)系考察精密取上述對照品溶液分別稀釋成13.1、26.2、52.4、104.8、209.6、419.2g/mL的對照品溶液,注入高效液相色譜儀,按上述色譜條件測定峰面積;以峰面積積分值為縱坐標(biāo),以橙皮苷進樣量(mg)為橫坐標(biāo)作線性回歸,繪制標(biāo)準(zhǔn)曲線,得回歸方程:Y=1471.42326X-10.75587(r=0.99974),結(jié)果表明,橙皮苷在0.131~4.192mg之間呈良好線性關(guān)系。
2.2.6精密度試驗按照測定方法,制備同一橙皮苷對照品溶液,精密吸取10μL重復(fù)進樣8次,測定其峰面積值。相對標(biāo)準(zhǔn)偏差(RSD)為0.03%。
2.2.7穩(wěn)定性試驗取141106批同一供試品溶液,設(shè)定分別在0、2、4、6、8、10h進樣10μL,按以上色譜條件進行測定。測得其峰面積值,RSD=0.252%,結(jié)果表明,在10h內(nèi)供試品溶液中的橙皮苷穩(wěn)定。
2.2.8重復(fù)性試驗取批號為141106(5g/袋,相當(dāng)于飲片4g)的健脾顆粒,供試品制備方法分別制備9份供試品溶液,測定橙皮苷含量,平均含量為1.1133mg/g,RSD為1.48%。
2.2.9加樣回收率試驗取已知含量的健脾顆粒樣品(批號:141106,橙皮苷含量:1.1133mg/g),添加橙皮苷對照品,按正文中含量測定方法測定橙皮苷含量,計算回收率,表明法有良好的回收率。結(jié)果見表1。
2.2.10樣品測定取6批健脾顆粒樣品分別按上述含量測定方法測定橙皮苷含量。測定結(jié)果見表2。
3討論
研究以黨參對照藥材作為對照,參照中國藥典一部黨參項下相關(guān)內(nèi)容進行本品TLC試驗。試驗結(jié)果:斑點模糊,色譜通道顏色較深,特征斑點不易檢視。又多次試驗其他提取方法和展開劑,結(jié)果:陰性有干擾。
在山楂的薄層鑒別中,參照藥典方法,以山楂對照藥材作對照,試用過多種提取方法和展開劑,進行薄層色譜試驗研究。結(jié)果:供試品色譜中均無明顯對應(yīng)斑點。
在麥芽薄層鑒別,以麥芽對照藥材作為對照,參照《中國藥典》一部麥芽項下相關(guān)方法進行本品TLC試驗。試驗結(jié)果:供試品色譜在與麥芽對照藥材的色譜相對應(yīng)的位置上顯相同顏色熒光斑點,陰性供試品無相對應(yīng)的斑點,但供試品色譜中對應(yīng)斑點不明顯,增加取樣量后,斑點無明顯改善,故此方法未采用。
參考文獻
[1] 國家藥典委員會.衛(wèi)生部藥品標(biāo)準(zhǔn)中藥成方制劑(第九冊).WS3-B-0381-90.
[2] 國家藥典委員會.中華人民共和國藥典(一部)[M].北京:中國醫(yī)藥科技出版社,2010.
[3] 國家藥典委員會.中華人民共和國藥典(四部)[M].北京:中國醫(yī)藥科技出版社,2015.
(編輯:穆麗華)
