一測多評法同時測定北青龍衣藥材中胡桃醌和胡桃酮的含量Δ
2017-03-03 06:45:13霍金海張海燕王偉明黑龍江省中醫藥科學院中藥研究所哈爾濱50036黑龍江省林下經濟資源研發與利用協同創新中心哈爾濱50040
中國藥房 2017年3期
關鍵詞:質量
霍金海,張海燕,王偉明,2#(.黑龍江省中醫藥科學院中藥研究所,哈爾濱 50036;2.黑龍江省林下經濟資源研發與利用協同創新中心,哈爾濱 50040)
一測多評法同時測定北青龍衣藥材中胡桃醌和胡桃酮的含量Δ
霍金海1*,張海燕1,王偉明1,2#(1.黑龍江省中醫藥科學院中藥研究所,哈爾濱 150036;2.黑龍江省林下經濟資源研發與利用協同創新中心,哈爾濱 150040)
目的:建立同時測定北青龍衣藥材中胡桃醌、胡桃酮含量的方法。方法:采用高效液相色譜法,以胡桃醌為內參物,計算其對胡桃酮的相對校正因子(RCF),通過RCF計算北青龍衣藥材中胡桃酮的含量;以外標法測定的胡桃酮含量作實測值,采用夾角余弦法比較一測多評法的計算值與實測值的差異。結果:胡桃醌、胡桃酮檢測質量濃度線性范圍分別為7.6~76.0 μg/mL(r=0.999 0)、8.2~82.0 μg/mL(r=0.999 4);精密度、穩定性、重復性試驗的RSD<3.0%;加樣回收率分別為95.37%~97.94%(RSD=1.04%,n=6)、99.13%~100.10%(RSD=0.33%,n=6)。計算值與實測值之間的夾角余弦值為0.999 84,兩者差異無統計學意義(P>0.05)。結論:該方法操作簡便,精密度、穩定性、重復性好,可用于北青龍衣藥材中胡桃醌、胡桃酮含量的同時測定。
北青龍衣;一測多評;高效液相色譜法;校正因子;胡桃醌;胡桃酮;含量
北青龍衣為胡桃科胡桃屬植物核桃楸Juglans mandshurica Maxim.的未成熟外果皮,主產于東北三省,以黑龍江省資源蘊藏量最大。萘醌類為公認的北青龍衣抗腫瘤主要成分,文獻表明從胡桃屬植物中已分離獲得60多種萘醌類化合物[1],其中以胡桃醌、胡桃酮為主要活性成分。目前國內外學者多集中于胡桃醌的含量測定方法研究,但胡桃醌化學性質不穩定,致使測定結果不理想。筆者前期研究發現,北青龍衣鮮品中胡桃醌含量較高,在干燥、貯存過程中能夠轉化成胡桃酮,二者總和相對穩定。因此,同時測定北青龍衣藥材中胡桃醌、胡桃酮的總量,對于北青龍衣藥材質量控制及品質評價具有實際意義。
目前,胡桃醌有化學合成對照品,而胡桃酮僅有少數植物化學家從胡桃屬植物中分離獲得,難以商品化,筆者也未見其含量測定方法的報道。近年來,一測多評技術采用一個對照品實現對多個成分的同步測定,對于一些制備困難或價格昂貴的對照品具有實際應用價值。該方法在白芍[2]、木香[3]等藥材中得到發展和驗證,黃連的一測多評標準已被2010年版《中國藥典》(一部)采納[4]。2015年版《中國藥典》(一部)繼續收錄了丹參等品種的一測多評含量測定方法[5]。本試驗針對北青龍衣藥材中主要指標成分胡桃醌、胡桃酮建立一測多評方法,解決了難以獲得胡桃酮對照品的困擾,為北青龍衣藥材的質量控制及采收期的確定奠定基礎。
1 材料
1.1 儀器
1100型高效液相色譜(HPLC)儀,包括G1315B DAD二級管陣列檢測器(美國Agilent公司);2010型HPLC儀,包括紫外雙波長檢測器(日本Shimadzu公司);KQ-300DB型數控超聲儀(昆山市超聲儀器有限公司,功率:300 W,頻率:40 kHz);BP211D型電子分析天平、BSA224S-CW型電子分析天平(德國Sartorius公司)。
1.2 試劑
胡桃醌對照品(成都瑞芬思生物科技有限公司,批號:H-075-131230,純度:>99%);胡桃酮對照品(筆者自制,經氫譜、碳譜檢測結構,HPLC面積歸一化法測定純度:>98%);磷酸、甲醇為色譜純,其余試劑均為分析純,水為超純水。
1.3 藥材
北青龍衣藥材于2014年7—8月采自黑龍江省不同產地(見表1),經黑龍江省中醫藥科學院初東君主任藥師鑒定為真品。剝取外果皮,通風處陰干,7 d后用于樣品制備(含水量均符合小于10%的要求)。
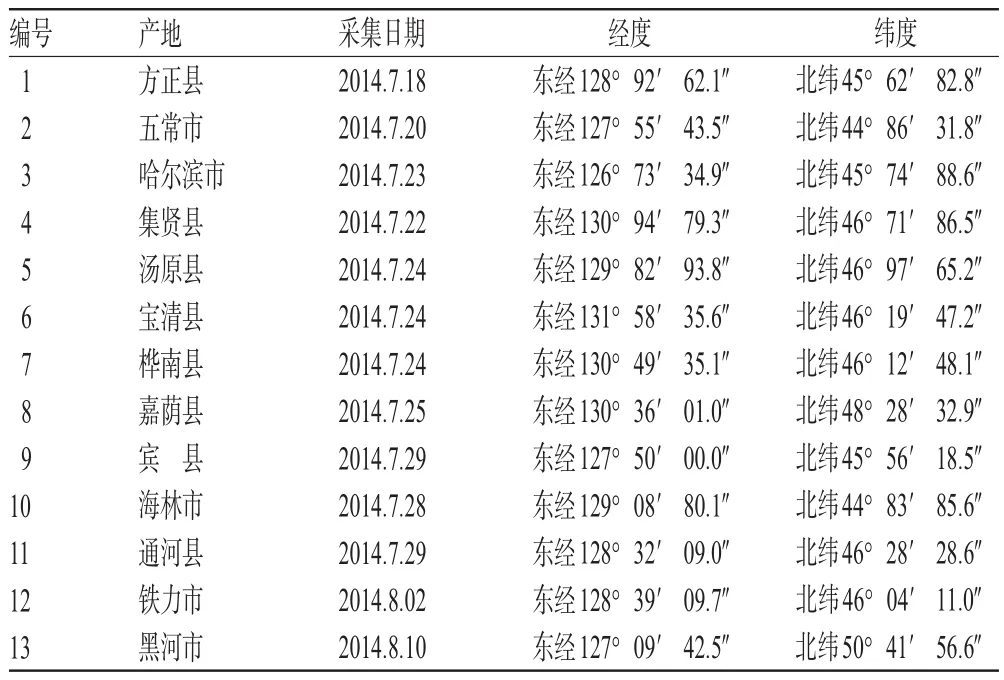
表1 北青龍衣藥材來源Tab 1 Source of J.mandshurica
2 方法與結果
2.1 原理
在一定線性范圍,成分的量(質量或濃度)與檢測器響應值成正比。在多指標(s,a,b,…,i,…)質量評價時,以藥材/成藥中某一典型有效成分作內參物(s),建立內參物對其他待測成分(a,b,…,i,…)間的相對校正因子(RCF),按下式計算:

式中,As為內參物對照品s峰面積,cs為內參物對照品s濃度,Ai為待測成分對照品i峰面積,ci為待測成分對照品i濃度。
在方法學建立時,主要是求出內參物與各待測成分間的RCF,并將其作為一個常數用于含量測定中。
應用RCF(fsa,fsb,fsc,…),結合內參物(s)實測值cs,計算待測成分(a,b,…,i,…)的濃度:

2.2 色譜條件與系統適用性試驗
色譜柱:Diamonsil C18(250 mm×4.6 mm,5 μm);流動相:甲醇-水(45∶55,V/V);流速:1.0 mL/min;檢測波長:265 nm;柱溫:30℃;進樣量:10μL。在上述色譜條件下,理論板數以胡桃酮、胡桃醌峰計均>3 000;各成分基線分離良好,分離度>1.5,詳見圖1。
2.3 溶液的制備
2.3.1 混合對照品溶液 取待測成分對照品適量,精密稱定,置于10 mL量瓶中,加甲醇溶解并定容,制成胡桃醌、胡桃酮質量濃度分別為0.076、0.082 mg/mL的混合對照品溶液。
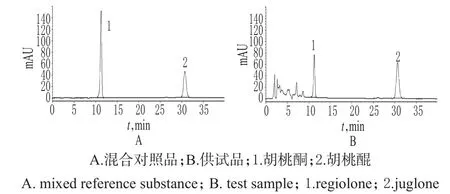
圖1 高效液相色譜圖Fig 1 HPLC chromatograms
2.3.2 供試品溶液 取樣品粉末(過60目篩)1 g,精密稱定,加甲醇25 mL,稱定質量,超聲處理30 min,放冷,再次稱定質量,加甲醇補足減失的質量,搖勻,經微孔濾膜濾過,取續濾液,即得。
2.4 線性關系考察
分別精密量取“2.3.1”項下混合對照品溶液1、2、3、5、10 mL,分別置于10 mL量瓶中,加流動相定容,制成系列混合對照品溶液。精密量取上述系列混合對照品溶液各10 μL,按“2.2”項下色譜條件進樣測定,記錄峰面積。以待測成分質量濃度(x,μg/mL)為橫坐標、峰面積(y)為縱坐標進行線性回歸,得胡桃醌、胡桃酮回歸方程分別為y=32.22x+79.04(r=0.999 0)、y=33.04x+ 111.3(r=0.999 4)。結果表明,胡桃醌、胡桃酮檢測質量濃度線性范圍分別為7.6~76.0、8.2~82.0 μg/mL。
2.5 精密度試驗
取“2.3.1”項下混合對照品溶液適量,按“2.2”項下色譜條件連續進樣測定6次,記錄峰面積。結果,胡桃醌、胡桃酮峰面積的RSD分別為0.98%、0.95%(n=6),表明儀器精密度良好。
2.6 穩定性試驗
取“2.3.2”項下供試品溶液(編號:1)適量,分別于室溫下放置0、2、4、6、8、10、12 h時按“2.2”項下色譜條件進樣測定,記錄峰面積。結果,胡桃醌、胡桃酮峰面積的RSD分別為0.51%、2.85%(n=7),表明供試品溶液在室溫放置12 h內基本穩定。
2.7 重復性試驗
精密稱取同一批樣品(編號:1)適量,按“2.3.2”項下方法制備供試品溶液,共6份,再按“2.2”項下色譜條件進樣測定,記錄峰面積并計算含量。結果,胡桃醌、胡桃酮平均含量分別為0.462 6、1.956 1 mg/g,RSD分別為2.91%、2.55%(n=6),表明本方法重復性良好。
2.8 加樣回收率試驗
取已知含量樣品(編號:1)適量,共6份,分別加入一定質量的待測成分對照品,按“2.3.2”項下方法制備供試品溶液,再按“2.2”項下色譜條件進樣測定,記錄峰面積并計算加樣回收率,結果見表2。
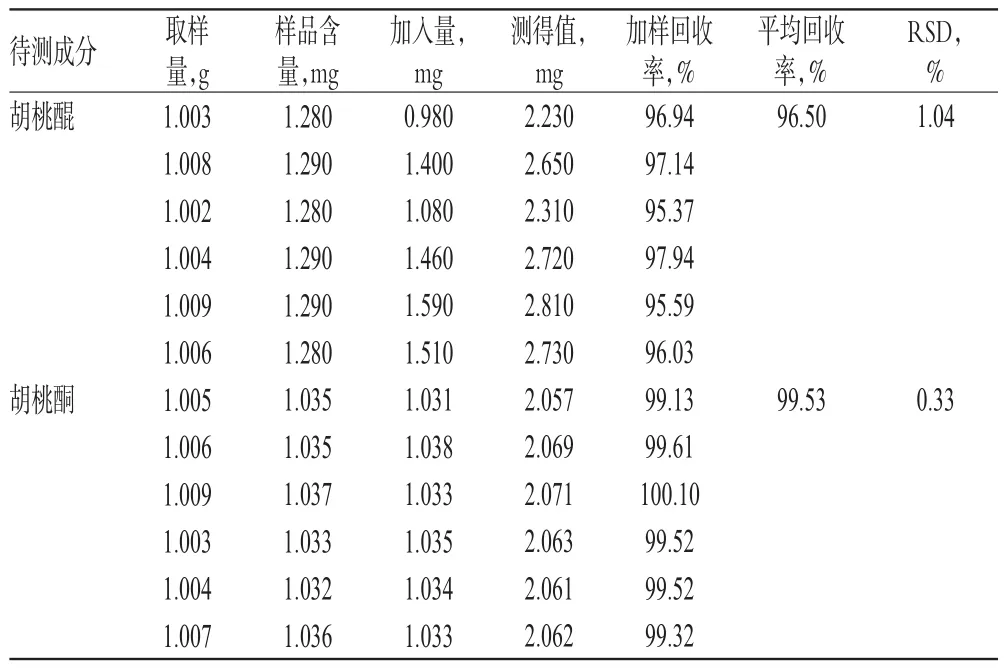
表2 加樣回收率試驗(n=6)Tab 2 Results of recovery test(n=6)
2.9 RCF的計算
以胡桃醌為內參物,按“2.1”項下公式(1)計算,結合“2.4”項下系列混合對照品溶液所得峰面積數據,計算胡桃醌對胡桃酮的RCF。結果,胡桃醌對胡桃酮的RCF為1.131。
2.10 RCF耐用性考察
2.1 0.1 色譜條件考察 取“2.3.1”項下混合對照品溶液,分別考察不同流動相[甲醇-0.2%磷酸溶液(55∶45,V/V)、甲醇-水(45∶55,V/V)]、不同流動相比例[甲醇-水(45∶55,V/V)、(50∶50,V/V)]、不同柱溫(20、30、40℃ )、不同流速(0.8、1.0、1.2 mL/min)對RCF的影響,詳見表3~表6。

表3 不同流動相對RCF的影響(n=6)Tab 3 Effect of different mobile phases on RCF(n=6)

表4 不同流動相比例對RCF的影響(n=6)Tab 4 Effect of different mobile phase ratios on RCF(n=6)
2.1 0.2 色譜柱和HPLC儀考察 取“2.3.1”項下混合對照品溶液,分別考察不同HPLC儀(安捷倫1100、島津2010)和不同色譜柱[Diamonsil C18(250 mm×4.6 mm,5 μm)、Kromasil 100-5C18(250 mm×4.6 mm,5 μm)、Agilent Extend C18(250 mm×4.6 mm,5 μm)3種色譜柱,詳見表7。
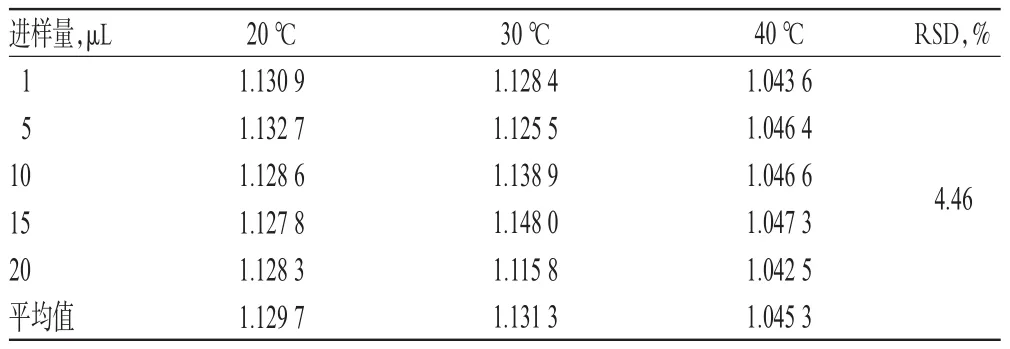
表5 不同柱溫對RCF的影響(n=6)Tab 5 Effect of different column temperature on RCF(n=6)
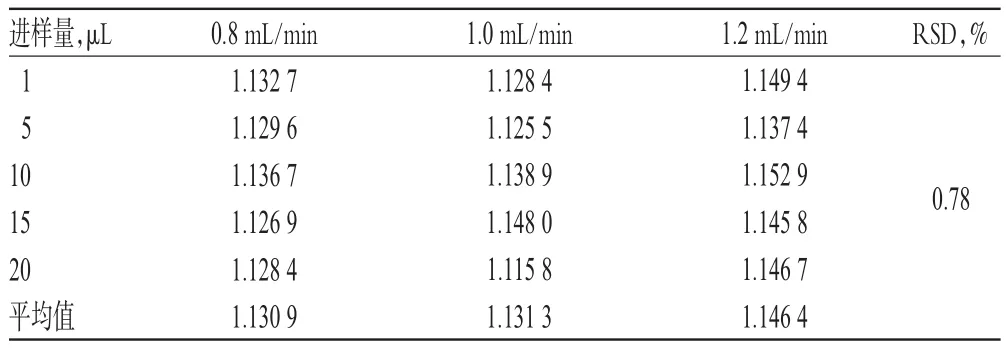
表6 不同流速對RCF的影響(n=6)Tab 6 Effect of different velocities on RCF(n=6)
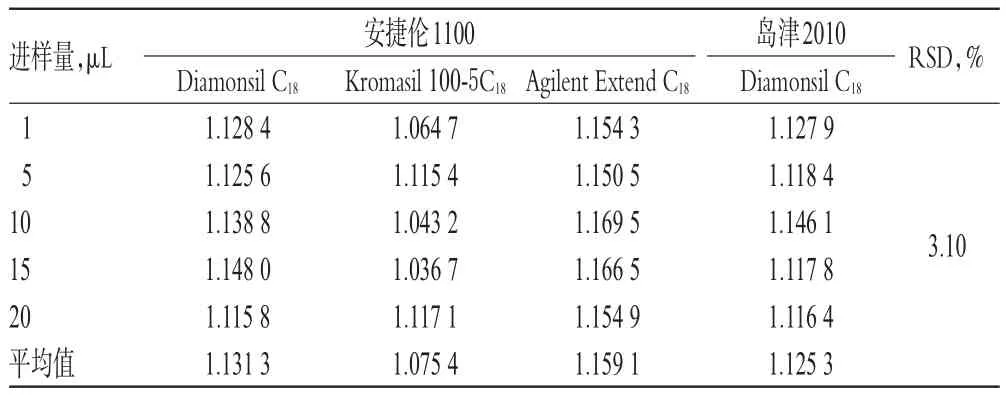
表7 不同儀器和色譜柱對RCF的影響(n=6)Tab 7 Effect of different instruments and chromatographic columns on RCF(n=6)
2.1 0.3 實驗室考察 取“2.3.1”項下混合對照品溶液考察3個不同實驗室對RCF的影響,詳見表8。
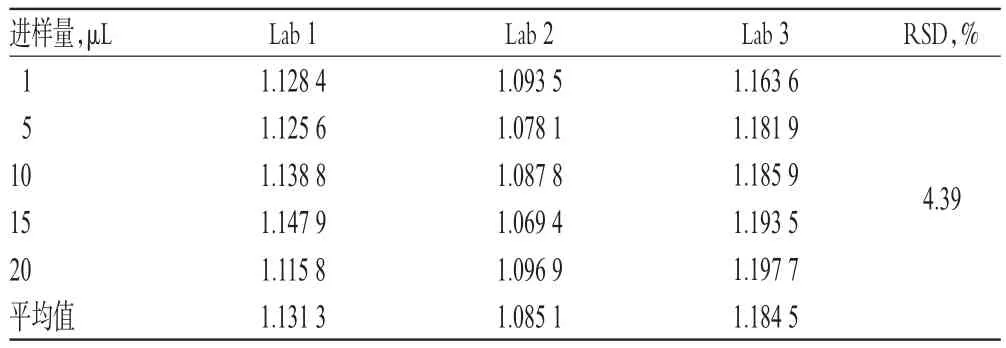
表8 不同實驗室對RCF的影響(n=6)Tab 8 Effect of different laboratories on RCF(n=6)
以上研究表明,該一測多評方法在流動相組成及比例、柱溫、流速發生小幅度變化,以及更換色譜柱、HPLC色譜儀、實驗室等條件下均具有較好耐用性,RSD均小于5%。
2.11 樣品含量測定
取表1中的樣品各適量,分別按“2.3.2”項下方法制備供試品溶液,再按“2.2”項下色譜條件進樣測定,記錄峰面積并計算樣品含量,見表9。采用一測多評法和外標法測得的胡桃酮的計算值和實測值之間的夾角余弦值為0.999 84,表明兩種方法所得含量差異無統計學意義(P>0.05)。
3 討論
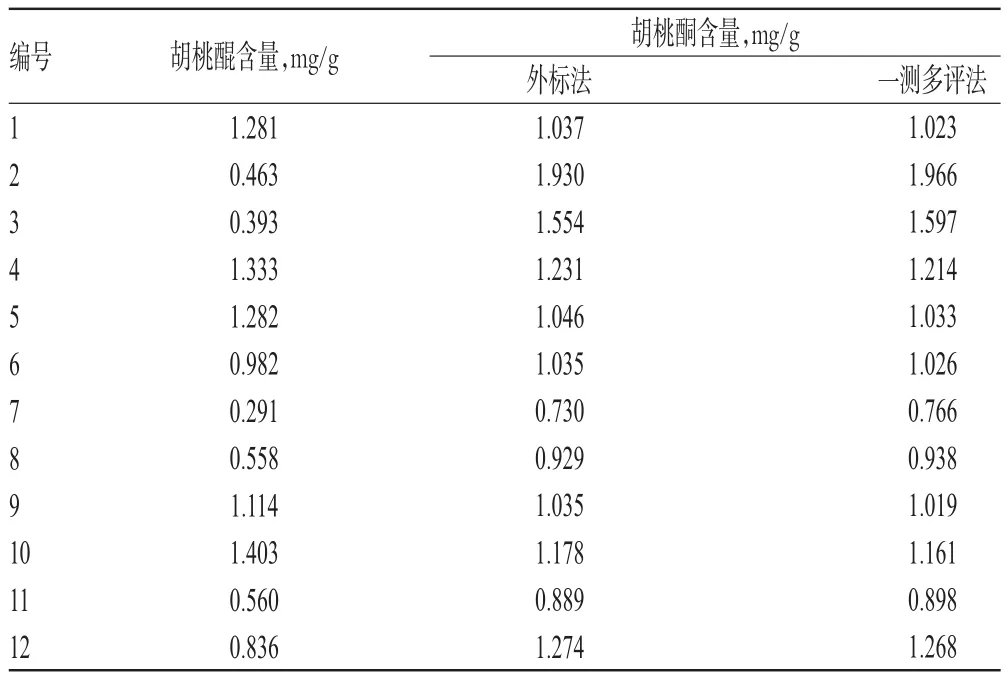
表9 樣品含量測定結果(n=6)Tab 9 Results of contents determination of samples(n=6)
筆者對不同流動相pH、流動相組成比例、柱溫、流速和檢測波長等因素進行了考察,結果RCF均無較大偏離(RSD<5%);通過方法學、方法的耐用性、系統適用性考察對一測多評法進行驗證,結果一測多評法計算值與外標法實測值差異無統計學意義(P>0.05),表明試驗建立的RCF準確可信。
預試驗中還檢測了北青龍衣鮮、干品中胡桃醌和胡桃酮含量,發現鮮品中無胡桃酮,且胡桃醌含量較高;然而鮮品中胡桃醌含量與干品中胡桃醌、胡桃酮含量之和相當;可見北青龍衣鮮、干品中胡桃醌、胡桃酮含量總和相對穩定。而單一測定胡桃醌的含量,受干燥方法和貯存時間的限制,難以準確反映北青龍衣藥材真實質量。
綜上所述,一測多評方法不僅能夠準確計算胡桃酮成分含量,并且實現了在對照品短缺的情況下,多指標成分的含量測定,為北青龍衣藥材的質量控制研究提供了更加科學準確的研究方法。
[1] Sun JX,Zhao XY,Fu XF,et al.Three new naphthalenyl glycosides from the root bark of Juglans cathayensis[J]. Chem Pharm Bull:Tokyo,2012,60(6):785-789.
[2] 黃山君,楊琪偉,石燕紅,等.一測多評法測定白芍中芍藥苷與芍藥內脂苷的含量[J].中國中藥雜志,2011,36(6):780-783.
[3] 楊憲,劉筱琴,張雪,等.HPLC一測多評法用于木香及3種含木香中藥復方制劑的質量評價[J].中國藥房,2014,25(11):1030-1034.
[4] 國家藥典委員會.中華人民共和國藥典:一部[S].2010年版.北京:中國醫藥科技出版社,2010:285.
[5] 國家藥典委員會.中華人民共和國藥典:一部[S].2015年
版.北京:中國醫藥科技出版社,2015:76.
Simultaneous Determination of Contents of Juglone and Regiolone in Juglans mandshurica by Quantitative Analysis of Multi-components by Single-maker
HUO Jinhai1,ZHANG Haiyan1,WANG Weiming1,2(1.Institute of Chinese Medicine,Heilongjiang Academy of Traditional Chinese Medicine,Harbin 150036,China;2.Heilongjiang Forest Economy Collaborative R&D and Innovation Center Resource Utilization,Harbin 150040,China)
OBJECTIVE:To establish a method for the simultaneous determination of juglone and regiolone in Juglans mandshurica.METHODS:HPLC was used and regiolone was used as internal reference,the relative correction factor(RCF)of regiolone and juglone wascalculated,then calculated the content of juglone based on RCF;the juglone content was used as measured value,which was determined by external standard method,angle cosine method was used to compared the difference of calculated value in quantitative analysis of multi-components by single-maker and measured value in external standard method.RESULTS:The lin-ear range was 7.6-76.0 μg/mL for juglone(r=0.999 0)and 8.2-82.0 μg/mL for regiolone(r=0.999 4);RSDs of precision,stability and reproducubility tests were lower than 3.0%;recoveries were 95.37%-97.94%(RSD=1.04%,n=6)and 99.13%-100.10%(RSD=0.33%,n=6).The cosine of the included angle between the calculated and measured values was 0.999 84,and there was no significant difference(P>0.05).CONCLUSIONS:The method is simple with good precision,stability and reproducibility,and can be used for the simultaneous determination of juglone and regiolone in J.mandshurica.
Juglans mandshurica;Quantitative analysis of multi-components by single-maker;HPLC;Correction factor;Juglone;Regiolone;Content
R284.1
A
1001-0408(2017)03-0380-04
2016-02-06
2016-05-16)
(編輯:張 靜)
國家自然科學基金資助項目(No.81503348);國家“重大新藥創制”科技重大專項課題(No.2009ZX09102-138)
*副研究員。研究方向:中藥品質評價及藥效物質基礎。電話:0451-55653086。E-mail:jinhaihuo@126.com
#通信作者:研究員。研究方向:中藥新藥研發。電話:0451-55665478。E-mail:zyyjy@163.com
DOI10.6039/j.issn.1001-0408.2017.03.26
猜你喜歡
中學生數理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(2018年6期)2018-04-22 03:16:54
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
