HPLC法測定二對甲苯磺酸緣生替尼原料藥中的有關物質
2017-03-03 06:45:20李家春李瑛光王振中黃文哲江蘇康緣藥業股份有限公司江蘇連云港222001
中國藥房 2017年3期
仲 艷,李家春,李瑛光,王振中,黃文哲,蕭 偉(江蘇康緣藥業股份有限公司,江蘇連云港 222001)
HPLC法測定二對甲苯磺酸緣生替尼原料藥中的有關物質
仲 艷*,李家春,李瑛光,王振中,黃文哲,蕭 偉#(江蘇康緣藥業股份有限公司,江蘇連云港 222001)
目的:建立測定二對甲苯磺酸緣生替尼原料藥中有關物質的方法。方法:采用高效液相色譜法。色譜柱為Waters Symmetry C18,流動相為甲醇-0.01 mol/L乙酸銨溶液(梯度洗脫),流速為1.0 mL/min,檢測波長為240 nm,柱溫為40℃,進樣量為10μL。結果:在該色譜條件下,主成分峰與各雜質峰分離度均良好;雜質A、B、C和二對甲苯磺酸緣生替尼檢測質量濃度線性范圍均為0.25~2.0μg/mL(r≥0.999 0),雜質A、B、C的定量限分別為0.5、0.5、2.5 ng;精密度、穩定性、重復性試驗的RSD<1.0%;加樣回收率分別為97.9%~102.6%、95.1%~107.7%、95.8%~107.5%,RSD分別為1.4%、4.2%、4.1%(n=9)。結論:該方法專屬性好、操作簡便,可用于二對甲苯磺酸緣生替尼原料藥中有關物質的測定。
二對甲苯磺酸緣生替尼原料藥;高效液相色譜法;有關物質
乳腺癌是女性最常見的惡性腫瘤,占女性所有惡性腫瘤的23%[1-3]。在我國,乳腺癌發病率逐年增加,已經成為城市女性的頭號癌癥殺手。二甲苯磺酸拉帕替尼是葛蘭素史克公司研發的一種新型的小分子靶向雙重酪氨酸激酶抑制劑,于2007年3月13日獲美國食品與藥品監督管理局批準上市,與抗癌藥物卡培他濱聯合用于治療晚期或轉移性表皮生長因子受體陽性乳腺癌。近年來,關于二甲苯磺酸拉帕替尼在其他類型腫瘤方面的研究越來越多,其有望成為一種潛在的多腫瘤靶向治療藥物[4-8]。二對甲苯磺酸緣生替尼是我公司以二甲苯磺酸拉帕替尼為先導化合物自主研發的新藥。由于本品在合成過程中難免引入合成原料、中間體、副產物等雜質,如6-碘喹唑啉-4-酮(雜質A)、3-氯-4-[(3-氟苯基)甲氧基]苯胺(雜質B)、N-[3-氯-4-(3-氟芐氧基)苯基]-6-碘-4-喹唑啉胺(雜質C)等,因此需要建立合適的測定方法以實現對本品中有關物質的控制。本研究依據2015年版《中國藥典》中藥品質量標準分析方法驗證指導原則的相關規定[9],參考相關文獻[10-12],優化高效液相色譜(HPLC)條件,建立了測定二對甲苯磺酸緣生替尼原料藥中有關物質的方法。
1 材料
1.1 儀器
1100型HPLC儀,包括G1329B二極管陣列檢測器、G1316A自動進樣器等(美國Agilent公司);BP211D型電子分析天平、PB-10型pH計(德國賽多利斯公司);ZMQS50001型超純水機(美國Millipore公司)。
1.2 藥品與試劑
二對甲苯磺酸緣生替尼對照品(批號:120701,純度:99.8%)和雜質A、B、C對照品(批號:120803、120811、120824,純度均為99.5%)及二對甲苯磺酸緣生替尼原料藥(批號:121001、121002、121003)均由江蘇康緣藥業股份有限公司提供;甲醇、乙腈為色譜純,其他試劑均為分析純,水為超純水。
2 方法
2.1 色譜條件
色譜柱:Waters Symmetry C18(250 mm×4.6 mm,5 μm);流動相:甲醇(A)-0.01 mol/L乙酸銨溶液(B),梯度洗脫(洗脫程序見表1);流速:1.0 mL/min;檢測波長:240 nm;柱溫:40℃;進樣量:10μL。

表1 梯度洗脫程序Tab 1 Gradient elution program
2.2 溶液的制備
2.2.1 空白對照溶液 以甲醇-水(50∶50,V/V)作空白對照溶液。
2.2.2 雜質對照品溶液 分別取雜質A、B、C對照品各約10 mg,精密稱定,置于同一10 mL量瓶中,加甲醇-乙腈溶液(50∶50,V/V)溶解并稀釋至刻度,搖勻,作為雜質對照品貯備液。精密量取該貯備液適量,加甲醇-水(50∶50,V/V)制成每1 mL中約含雜質A、B、C各1 μg的雜質對照品溶液。
2.2.3 供試品溶液 取樣品適量,精密稱定,加甲醇-水(50∶50,V/V)溶解并制成每1 mL中約含二對甲苯磺酸緣生替尼1 mg的供試品溶液。
2.2.4 對照溶液 精密量取“2.2.3”項下供試品溶液1 mL,置于100 mL量瓶中,加甲醇-水(50∶50,V/V)稀釋至刻度,搖勻,即得對照溶液。
2.2.5 系統適用性溶液 取二對甲苯磺酸緣生替尼對照品約20 mg,精密稱定,置于20 mL量瓶中,精密加入“2.2.2”項下的雜質對照品貯備液0.2 mL,再加甲醇-水(50∶50,V/V)溶解并稀釋制成每1 mL中約含二對甲苯磺酸緣生替尼和雜質A、B、C分別為1.10 mg、10.67 μg、10.64 μg、10.19 μg的系統適用性溶液。
2.3 專屬性試驗
2.3.1 系統適用性試驗 精密量取“2.2”項下空白對照溶液、供試品溶液、系統適用性溶液各10μL注入HPLC儀進樣,記錄色譜,詳見圖1。結果表明,空白對照對樣品中有關物質測定無干擾,二對甲苯磺酸、雜質A、雜質B、緣生替尼、雜質C的保留時間分別是4.242、15.519、22.335、30.164、43.565 min,主成分峰與各雜質峰的分離度良好,各雜質峰間的分離度均>1.5,拖尾因子均在0.9~1.1范圍內。
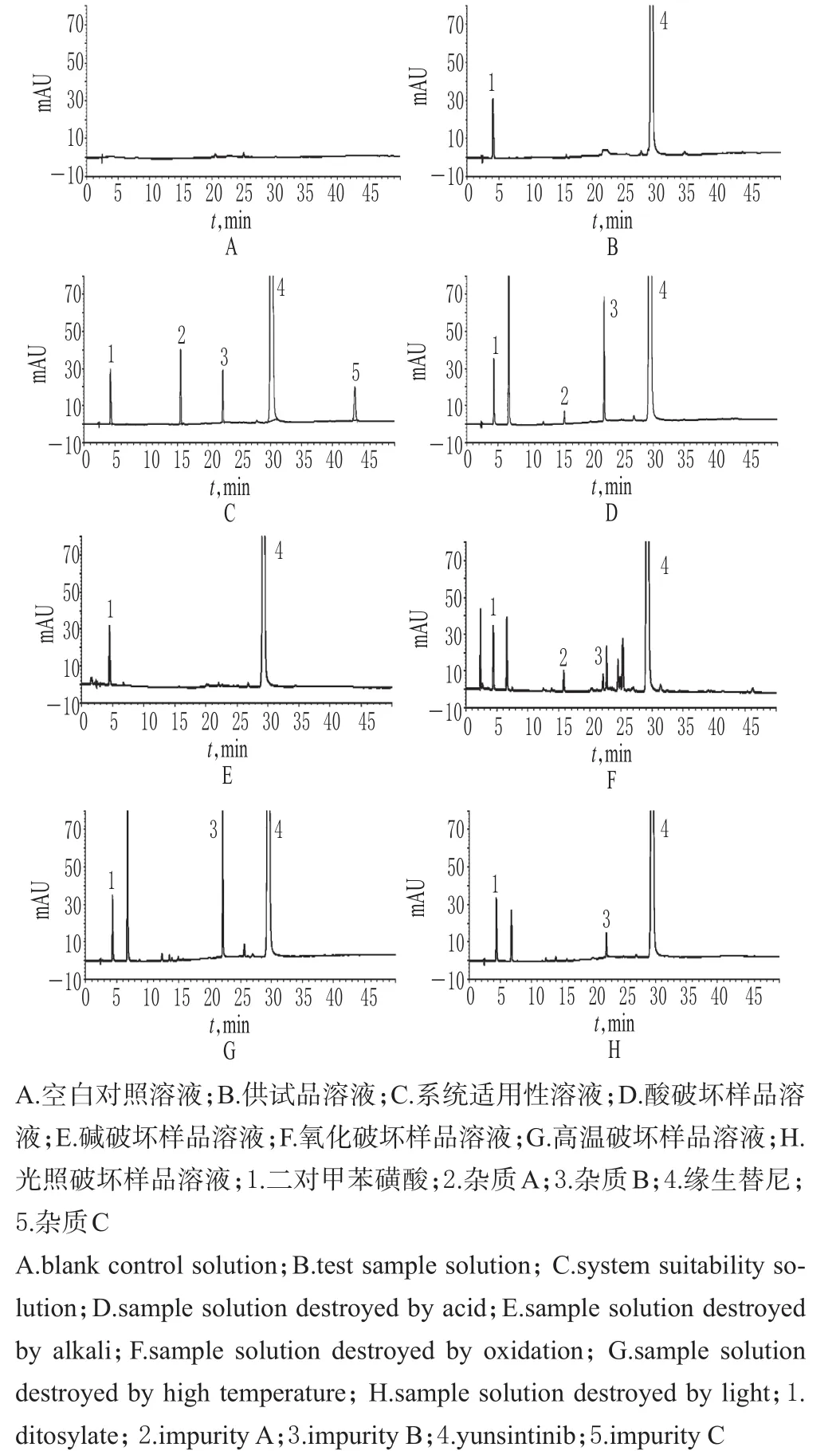
圖1 高效液相色譜圖Fig 1 HPLC chromatograms
2.3.2 強制降解試驗 分別精密稱取樣品約50 mg,共5份,各置于50 mL量瓶中,一份加1 mol/L鹽酸溶液10 mL,在水浴(92℃)中加熱4 h,放冷,用1 mol/L氫氧化鈉溶液調節pH至中性,加甲醇-水(50∶50,V/V)稀釋至刻度,搖勻,作為酸破壞樣品溶液;一份加1 mol/L氫氧化鈉溶液10 mL,在水浴(92℃)中加熱8 h,放冷,用1 mol/L鹽酸溶液調pH至中性,加甲醇-水(50∶50,V/V)稀釋至刻度,搖勻,作為堿破壞樣品溶液;一份加0.3%過氧化氫溶液10 mL,在水浴(92℃)下加熱0.5 h,放冷,加甲醇-水(50∶50,V/V)稀釋至刻度,搖勻,作為氧化破壞樣品溶液;一份加甲醇-水(50∶50,V/V)10 mL,在水浴(92℃)下加熱10 h,放冷,加甲醇-水(50∶50,V/V)稀釋至刻度,搖勻,作為高溫破壞樣品溶液;一份加甲醇-水(50∶50,V/V)溶解并稀釋至刻度,在強光4 500 lx下照射10 d,作為光照破壞樣品溶液。分別精密量取10μL上述5種破壞樣品溶液注入HPLC儀測定,記錄色譜,詳見圖1。結果表明,樣品在堿破壞條件下破壞不明顯;在酸、氧化、高溫、光照破壞條件下破壞明顯,且均能檢出雜質B;在酸及氧化破壞條件下均檢出雜質A。在該色譜條件下,主成分峰與各雜質峰、各雜質峰之間均可以達到良好的分離,證明本方法的專屬性良好,適宜進行樣品的有關物質測定。
2.4 線性關系考察
取雜質A、B、C及二對甲苯磺酸緣生替尼對照品各約10 mg,精密稱定,置于同一100 mL量瓶中,加甲醇-水(50∶50,V/V)溶解并稀釋至刻度,搖勻,作為線性工作貯備液。精密量取上述貯備液適量,分別加甲醇-水(50∶50,V/V)稀釋制成各成分質量濃度均約為0.25、0.5、1.0、1.5、2.0 μg/mL的系列線性工作溶液,按“2.1”項下色譜條件進樣測定,并以待測成分峰面積(y)為縱坐標、質量濃度(x,μg/mL)為橫坐標進行線性回歸,得到雜質A、B、C及二對甲苯磺酸緣生替尼的回歸方程和線性范圍,并用二對甲苯磺酸緣生替尼與雜質A、B、C的回歸方程的斜率相比得校正因子,詳見表2。
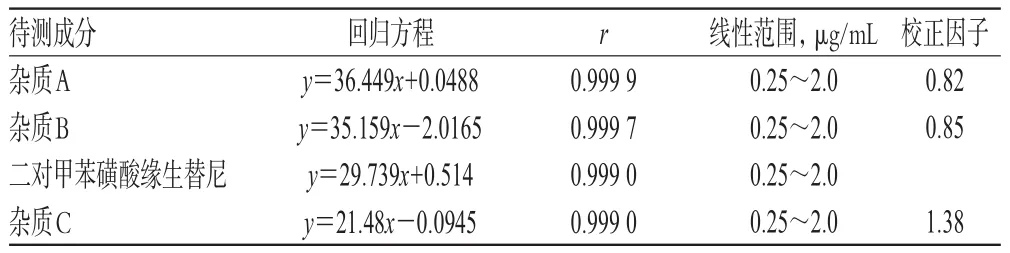
表2 回歸方程、線性范圍和校正因子Tab 2 Regression equations,linear range and correction factor
2.5 定量限與檢測限考察
取“2.4”項下線性工作貯備液適量,加空白對照溶液逐級稀釋至一定質量濃度,分別按“2.1”項下色譜條件進樣測定,記錄峰面積。結果,當信噪比為10∶1時計算得到雜質A、B、C及二對甲苯磺酸緣生替尼的定量限分別為0.5、0.5、2.5、1.0 ng;當信噪比為3∶1時計算得到雜質A、B、C及二對甲苯磺酸緣生替尼的檢測限分別為0.1、0.1、0.1、0.2 ng。
2.6 精密度試驗
取“2.4”項下雜質A、B、C及二對甲苯磺酸緣生替尼質量濃度分別為1.0μg/mL的線性工作溶液適量,按“2.1”項下色譜條件連續進樣測定6次,記錄峰面積。結果,雜質A、B、C及緣生替尼峰面積的RSD分別為0.2%、0.3%、0.3%、0.1%(n=6),表明儀器精密度良好。
2.7 穩定性試驗
精密稱取樣品(批號:121001)適量,分別按“2.2.2”和“2.2.3”項下方法制備供試品溶液和對照溶液。取上述兩種溶液各適量,分別于室溫放置0、2、4、6、8、12 h時按“2.1”項下色譜條件進樣測定,記錄峰面積。結果,對照溶液緣生替尼峰面積的RSD=0.5%(n=6),未知最大單個雜質及雜質總和峰面積的RSD均為0.1%(n=6),表明供試品溶液在室溫放置12 h內較為穩定。
2.8 重復性試驗
精密稱取樣品(批號:121001)適量,分別按“2.2.2”和“2.2.3”項下方法平行制備6份供試品溶液和6份對照溶液,分別按“2.1”項下色譜條件進樣測定,記錄峰面積。結果,未知最大單個雜質及雜質總和峰面積的RSD分別為0.1%和0.2%(n=6),表明本方法重復性良好。
2.9 加樣回收率試驗
精密稱取樣品(批號:121001)約10 mg,共9份,每3份分別加入約相當于二對甲苯磺酸緣生替尼量的0.05%、0.10%、0.15%的雜質A、B、C對照品,分別按“2.2.3”和“2.2.2”項下方法制備供試品溶液和雜質對照品溶液,按“2.1”項下色譜條件進樣測定并計算加樣回收率,結果見表3。
2.10 樣品有關物質測定
取3批樣品各適量,分別按“2.2.2”和“2.2.3”項下方法制備供試品溶液和對照溶液。精密量取對照溶液10 μL注入HPLC儀中,調節檢測靈敏度,使主成分色譜峰的峰高為滿量程的20%~25%;再精密量取上述兩種溶液各10μL,分別注入HPLC儀進樣測定,記錄峰面積,并以1%自身對照法計算樣品中有關物質的含量,結果見表4。
3 討論
3.1 檢測波長的選擇
因二對甲苯磺酸緣生替尼在240 nm波長處有最大吸收,且已知雜質A、B、C和強制降解產生的未知雜質均在240 nm波長處有較大吸收,綜合考慮選擇240 nm作為本方法的檢測波長。
3.2 色譜柱的選擇
通過對4個生產廠家的5根色譜柱進行考察——色譜柱1:Waters Symmetry C18(250 mm×4.6 mm,5 μm)、色譜柱2:Waters Symmetry Shield RP18 C18(250 mm× 4.6 mm,5 μm)、色譜柱3:Phenomenex Luna C18(250 mm×4.6 mm,5 μm)、色譜柱4:Welch Polar C18(250 mm×4.6 mm,5 μm)、色譜柱5:Kromasil 100-5C18(250 mm×4.6 mm,5 μm),結果表明,采用Waters Symmetry C18(250 mm×4.6 mm,5 μm)對破壞樣品溶液的分離度最佳,所以最終選擇該色譜柱進行試驗。
3.3 流動相的選擇
流動相采用甲醇-水或乙腈-水時,二對甲苯磺酸和緣生替尼均峰形差,因此考察了磷酸鉀鹽、磷酸銨鹽和乙酸銨鹽改善峰形。結果,3種無機鹽均能有效改善峰形,考慮到乙酸銨為揮發性無機鹽,對儀器系統的損害小,所以確定選擇乙酸銨。通過考察發現,乙酸銨在0.01 mol/L濃度下,強制降解試驗各破壞樣品的各色譜峰均可達到有效分離,所以乙酸銨濃度確定為0.01 mol/L。而以甲醇作為有機相時緣生替尼的色譜峰形較使用乙腈時好,所以有機相確定選擇甲醇。另外,采用等度洗脫時,二對甲苯磺酸色譜峰與強制降解產生的雜質峰難以分開,因此最終選擇甲醇-0.01 mol/L乙酸銨溶液為流動相梯度洗脫。
3.4 方法耐用性考察
在其他色譜條件不變情況下,當分別選擇柱溫變化±5℃、流速變化±0.2 mL/min、流動相中甲醇比例變化±5%、流動相中乙酸銨溶液濃度變化±50%時,考察緣生替尼峰與各雜質峰的分離情況,以驗證方法的耐用性。結果表明,上述各種條件下緣生替尼峰與各雜質峰、各雜質峰之間的分離度均符合規定,表明本方法耐用性良好。
3.5 強制降解試驗的物料平衡考察
在強制降解試驗中,酸、堿、氧化、高溫、光照破壞試驗的樣品物料平衡值均在95%~105%范圍內,表明該色譜條件可以準確地測定樣品中的有關物質。
3.6 有關物質限度的確定
本試驗采用1%自身對照法測定有關物質,因在強制降解試驗中,酸、氧化、高溫及光照破壞條件下均能使樣品降解出雜質B,且3批樣品中未知最大單個雜質的量均在0.1%以下,雜質總量均在0.3%以下。其限度暫定為:除溶劑和二對甲苯磺酸峰外,雜質B峰面積不得大于對照溶液主峰面積的0.1倍(即0.1%),其他單個雜質峰面積不得大于對照溶液主峰面積的0.2倍(即0.2%),各雜質峰面積的和不得大于對照溶液主峰面積的1.0倍(即1.0%)。
綜上所述,本方法專屬性好、操作簡便,可用于二對甲苯磺酸緣生替尼原料藥中有關物質的測定。
[1] 黃哲宇,陳萬青,吳春曉.中國女性乳腺癌的發病和死亡現況:全國32個腫瘤登記點2003—2007年資料分析報告[J].腫瘤,2012,32(6):435-439.
[2] 方瓊英,吳瓊,張秀玲.乳腺癌的流行現狀分析[J].中國社會醫學雜志,2012,29(5):333-335.
[3] 唐志柳,白潔,顧麗娜.2000—2010年我國前列腺癌和乳腺癌流行狀況的系統性綜述[J].中國腫瘤,2013,22(4):260-264.
[4] 婁瑩,黃韜.乳腺癌靶向治療進展[J].腫瘤學雜志,2009,15(9):788-791.
[5] 吳慧芳,王海琳,馮煥榮.拉帕替尼聯合順鉑對人卵巢癌細胞SKOV3生長的影響[J].中國現代醫藥雜志,2012,14(4):12-15.
[6] 何祥萌,張凌巖,李英.吉非替尼和拉帕替尼對HEL細胞增殖的抑制作用[J].中國實驗血液學雜志,2012,20(2):372-375.
[7] 陳偉,劉永梅.拉帕替尼:作用于表皮生長因子受體的靶向抗腫瘤新藥[J].藥品評價,2012,9(12):10-12.
[8] 馮煥榮,李青,王海琳.新型靶向治療藥物拉帕替尼的研究進展[J].現代生物醫學進展,2012,12(4):746-747.
[9] 國家藥典委員會.中華人民共和國藥典:二部[S].2015年版.北京:中國醫藥科技出版社,2015:374-377.
[10] 仲艷,陳保來,蘇倩倩.HPLC法測定二甲苯磺酸拉帕替尼含量及其有關物質[J].中國藥師,2013,16(11):1672-1675.
[11] 徐劍華.甲磺酸拉帕替尼有關物質的HPLC法測定[J].中國醫藥工業雜志,2015,46(10):1113-1116.
[12] 仲艷,傅小勤,李家春.HPLC高效液相色譜法測定二甲苯磺酸拉帕替尼有關物質[J].中國藥房,2013,24(37):3528-3530.
(編輯:周 箐)
Determination of Related Substances in Yunsintinib Ditosylate Active Pharmaceutical Ingredients by HPLC
ZHONG Yan,LI Jiachun,LI Yingguang,WANG Zhenzhong,HUANG Wenzhe,XIAO Wei(Jiangsu Kanion Pharmaceutical Co.,Ltd.,Jiangsu Lianyungang 222001,China)
OBJECTIVE:To establish a method for the determination of related substances in yunsintinib ditosylate active pharmaceutical ingredient.METHODS:HPLC was performed on the column of Waters Symmetry C18with mobile phase of methanol-0.01 mol/L ammonium acetate solution(gradient elution),flow rate was 1.0 mL/min,the detection wavelength was 240 nm,column temperature was 40℃,and injection volume was 10μL.RESULTS:Under the chromatographic conditions,the main peaks and each impurity peak were well separated;The linear range of impurity A,B,C and yunsintinib ditosylaie were 0.25-2.0 μg/mL(r≥0.999 0);the quantification limits of impurity A,B and C were 0.5,0.5 and 2.5 ng,respectively;RSDs of precision,stability and reproducibility tests were lower than 1.0%;recoveries were 97.9%-102.6%(RSD=1.4%,n=9),95.1%-107.7%(RSD=4.2%,n=9),95.8%-107.5%(RSD=4.1%,n=9),respectively.CONCLUSIONS:The method is specific and simple,and can be used for the determination of related substances in yunsintinib ditosylate active pharmaceutical ingredients.
Yunsintinib ditosylate active pharmaceutincal ingredients;HPLC;Related substances
R927
A
1001-0408(2017)03-0412-04
2016-02-03
2016-12-14)
*高級工程師。研究方向:藥物分析。E-mail:zy521-521@126. com
#通信作者:高級工程師,博士。研究方向:創新藥物的研發。電話:0518-81152337。E-mail:wzhzh-nj@163.net
DOI10.6039/j.issn.1001-0408.2017.03.35
