線性-樹枝狀共聚物載藥微球的制備、表征及體外釋放
2017-03-08 03:45:36楊李影戎宗明
華東理工大學學報(自然科學版) 2017年1期
楊李影, 郭 睿, 戎宗明
(1.華東理工大學化學與分子工程學院,上海 200237; 2.巴斯夫上海涂料有限公司,上海 201108)
線性-樹枝狀共聚物載藥微球的制備、表征及體外釋放
楊李影1, 郭 睿2, 戎宗明1
(1.華東理工大學化學與分子工程學院,上海 200237; 2.巴斯夫上海涂料有限公司,上海 201108)
以自制的生物可降解線性-樹枝狀兩嵌段兩親共聚物材料為藥物載體,采用透析法制備了雷帕霉素載藥微球。通過掃描電子顯微鏡(SEM)、動態光散射儀(DLS)、紫外分光光度計對雷帕霉素載藥微球的表面形貌、粒徑分布、載藥量及釋放行為進行了研究。結果表明:載藥微球的載藥量和包封率分別可達到40%和90%以上,與啞鈴型三嵌段兩親共聚物載藥微球相當,但平均粒徑大幅降低(粒徑約300 nm),同樣具有顯著的緩釋作用。
線性-樹枝狀共聚物; 生物可降解; 載藥微球; 體外釋放; 雷帕霉素
抗腫瘤藥物的控制釋放是惡性腫瘤化療的關鍵,而生物可降解材料成為主要的藥物載體[1-4],尤其是聚乙二醇-聚乳酸兩親型嵌段共聚物,由于其獨特的自組裝等優異功能,成為研究的重點并被廣泛地應用到載藥體系中[5-7]。
本課題組曾自制了生物可降解的聚L-丙交酯-dendron-聚乙二醇-dendron-聚L-丙交酯啞鈴型三嵌段兩親共聚物[8],實驗研究發現其在選擇性溶劑中能自組裝形成規整的空心微球[9],利用其自組裝的特點,采用透析法制備了載雷帕霉素微球,得到了載藥量和包封率分別可達40%和91%、平均粒徑在300~500 nm的載藥微球[10];對于在水中具有一定溶解度的替莫唑胺藥物,則以替莫唑胺飽和水溶液作選擇性溶劑,透析制得了載藥量可達40%、平均粒徑不大于800 nm的載藥微球[11]。這兩種載藥微球都具有緩釋功能,體外緩釋時間最長可達到900 h之久。
為了進一步減小載藥微球的粒徑,又以聚乙二醇單甲醚代替聚乙二醇作為起始劑,通過發散法合成了一系列線性-樹枝狀兩嵌段共聚物,并通過增加親水鏈長的方法來提高其所形成的膠束在水溶液中的穩定性[12]。本文將以此類嵌段共聚物為載體,進行包裹免疫抑制藥物雷帕霉素(Rapamycin,RAPA)的研究,在確定制備工藝的條件下,探究不同結構的線性-樹枝狀兩嵌段共聚物對其所形成的載藥微球的載藥性能,如粒徑分布、形態、緩釋等性能的影響,以獲得性能更為優異的藥物載體材料。
1 實驗部分
1.1 主要原料與儀器
主要原料:雷帕霉素(醫用級),上海東方醫院提供;透析袋,7 000 Du,VISKASE?公司;線性-樹枝狀共聚物(mPEG(5 000)-Gx-PLLA(y)n,x為2、3、4,y為2 500、5 000、10 000,n為4、8、16),實驗室自制,其結構如圖1所示,文中所用線性-樹枝狀兩嵌段共聚物的結構信息見表1。
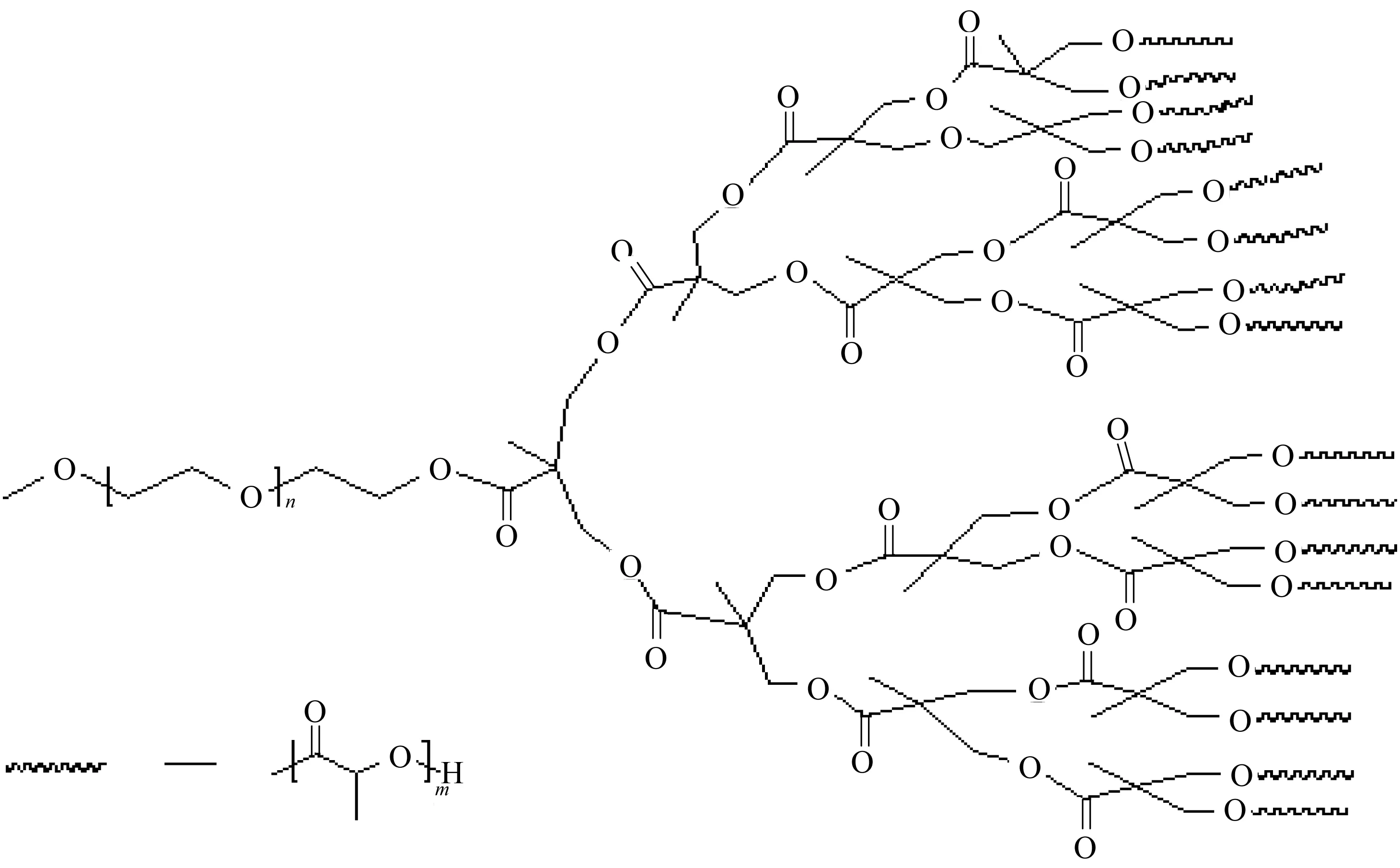
圖1 四代線性-樹枝狀共聚物結構
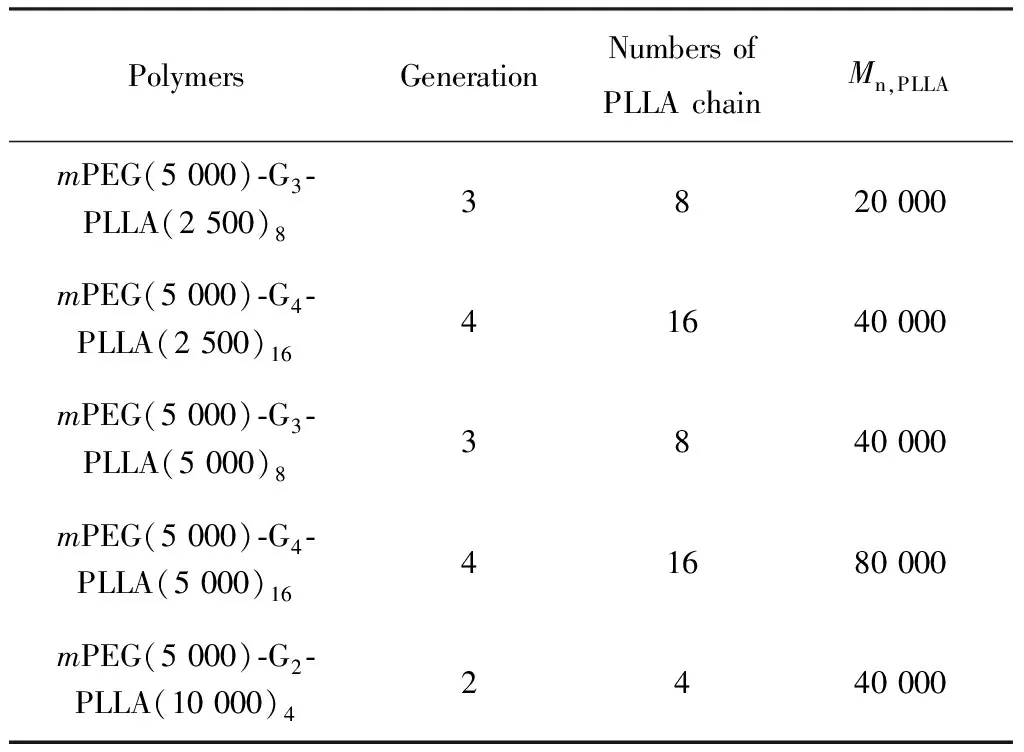
PolymersGenerationNumbersofPLLAchainMn,PLLAmPEG(5000)?G3?PLLA(2500)83820000mPEG(5000)?G4?PLLA(2500)1641640000mPEG(5000)?G3?PLLA(5000)83840000mPEG(5000)?G4?PLLA(5000)1641680000mPEG(5000)?G2?PLLA(10000)42440000
掃描電子顯微鏡(SEM,日本Hitachi S-3400);動態光散射(DLS,Zetasizer Nano,英國馬爾文儀器有限公司);紫外分光光度計(UV765,上海精密科技有限公司)。
1.2 雷帕霉素載藥微球的制備及表征
采用透析法制備聚合物載藥微球。稱取一定量的線性-樹枝狀共聚物及RAPA藥物粉末于四氫呋喃中,超聲待其完全溶解后,將混合液轉入透析袋內。在35 ℃恒溫下,在去離子水中透析,低速攪拌,間隔4 h更換外水相,直至溶劑除盡。待透析結束后,將透析袋內溶液高速離心40 min,取出底部白色沉淀,經冷凍干燥后得到固體載藥微球。空微球的制備方法與載藥微球的制備方法相似,僅在四氫呋喃溶劑中不加入雷帕霉素即可。
采用動態光散射儀(DLS)測量微球的平均粒徑及分散系數(PDI)。取一定體積的微球溶液,在25 ℃下,測量樣品粒徑大小及分布系數,每個樣品測量3次。采用掃描電鏡(SEM)觀察微球形貌特征。將微球溶液樣品分散在云母片上,自然干燥,做噴金處理后在掃描電鏡下觀察微球的形貌及大小。
1.3 載藥微球載藥量及包封率的測定
稱取一定質量冷凍干燥后的載雷帕霉素的微球粉末,使其完全溶解于一定量二氯甲烷中,用紫外分光光度計測得其在波長278 nm處的吸光度A。同時根據RAPA在二氯甲烷中的標準曲線計算得出載藥微球中RAPA的含量。載藥微球的實測載藥量(DL)及包封率(EE)計算公式:
(1)
(2)
式中:ms為包裹進微球的藥物質量;m0為總的共聚物投入質量;mt為總的藥物投入質量。
1.4 雷帕霉素載藥微球體外釋放測定方法
稱取一定質量的雷帕霉素,用乙醇溶解,按照乙醇與PBS緩沖液體積比1∶9,加入pH=7.4的PBS緩沖液,分別配成一系列不同濃度的溶液,用不含雷帕霉素的PBS與乙醇的混合液作空白參比,用紫外分光光度計測定其在278 nm處的吸光度(A),繪制得到標準曲線,A=51.96c-0.008 6,相關系數R為0.999 1,式中雷帕霉素質量濃度c的單位為mg/mL。
稱取一定質量的載雷帕霉素微球粉末分散在pH=7.4的PBS緩沖溶液中。將溶液轉移至透析袋內,浸置在37 ℃的PBS溶液中模擬體外釋放。每間隔一段時間,取2 mL的緩釋溶液,同時補加等量的新鮮PBS緩沖液,保持緩沖液的體積不變。采用紫外分光光度計測得緩釋溶液的吸光度,根據標準曲線方程計算出不同時間RAPA的累計釋放量,繪制體外RAPA累計釋放率-時間曲線。
2 結果與討論
2.1 載藥微球的載藥性能
2.1.1 最佳投藥質量比的確定 以mPEG(5 000)-G4-PLLA(2 500)16和mPEG(5 000)-G4-PLLA(5 000)16兩個共聚物制備載藥微球為例,考察了投藥質量比對載藥微球的載藥量、包封率、粒徑及形態的影響,實驗結果見表2和圖2。從表2中可以看出,隨投藥質量比的增加,兩種共聚物的載藥微球實測的載藥量呈線性增加,但載藥微球的平均粒徑變化不大;在相同的投藥比時,由于mPEG(5 000)-G4-PLLA(5 000)16的疏水鏈段PLLA較mPEG(5 000)-G4-PLLA(2 500)16增長了1倍,能吸附較多的藥物,因此其載藥量和包封率也相對較高,相應的微球粒徑也較大些。
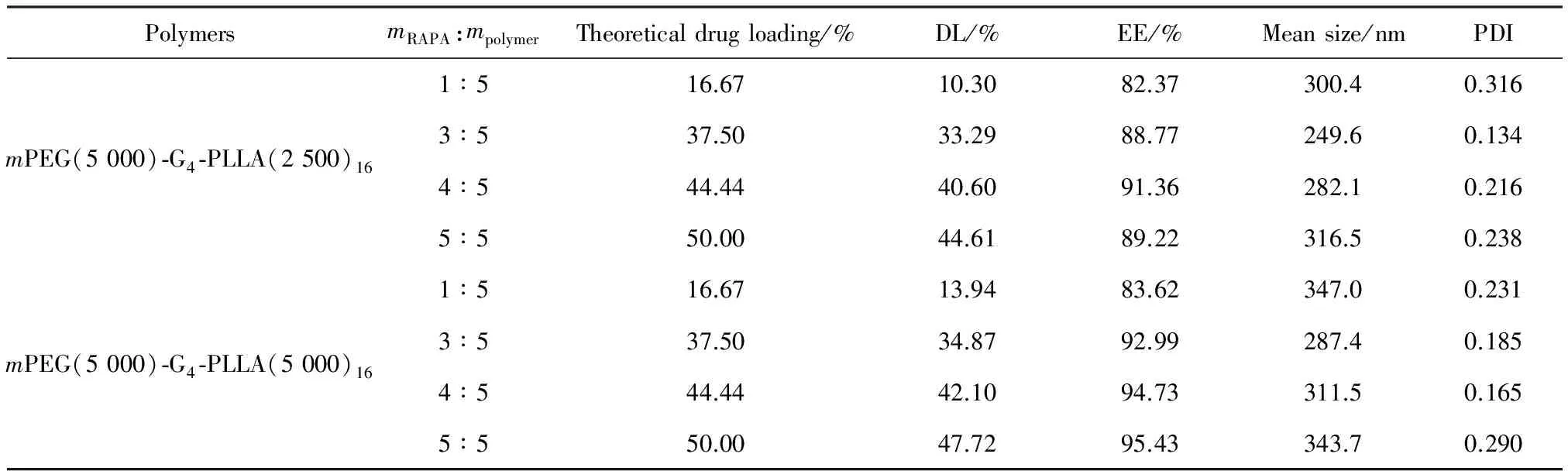
表2 RAPA不同投藥質量比對載藥微球平均粒徑、載藥量以及包封率的影響
為了進一步確定藥物是否完全被包裹進微球內部,采用掃描電鏡(SEM)觀察了所制備的載藥微球的形貌,結果見圖2,從圖2中可以看到載藥微球本身呈空心球形,且分散良好。對于mPEG(5 000)-G4-PLLA(2 500)16共聚物載藥微球,在投藥質量比為3∶5時,載藥微球表面光滑,見圖2(a);但在投藥質量比為4∶5(圖2(b))和5∶5(圖2(c))時,微球表面有類似藥物顆粒黏附,外面也可看到零星藥物顆粒存在,說明雷帕霉素藥物并沒有完全被包裹進微球內部,實驗測得的載藥量和包封率并不真實。因此,其最佳的投藥質量比應為3∶5。而mPEG(5 000)-G4-PLLA(5 000)16共聚物的載藥微球表面和外面均未見有藥物顆粒,見圖2(d~f),載藥量和包封率也隨著投藥比的增加而增大。考慮到mPEG(5 000)-G4-PLLA(5 000)16是該系列聚合物中聚乳酸含量最多的,為兼顧低代數的聚合物,其投藥質量比選擇為4∶5。

圖2 不同投藥質量比載藥微球的SEM圖
2.1.2 載藥微球的性能與聚合物分子結構間的關系 由于合成線性-樹枝狀一代共聚物中聚乙二醇的體積分數φEO>0.5,不能形成規整的球形膠束[12],故只考察了2~4代線性-樹枝狀共聚物的載藥微球在確定的投藥質量比時,載藥微球的載藥量、包封率和粒徑隨代數的變化關系,結果見表3。
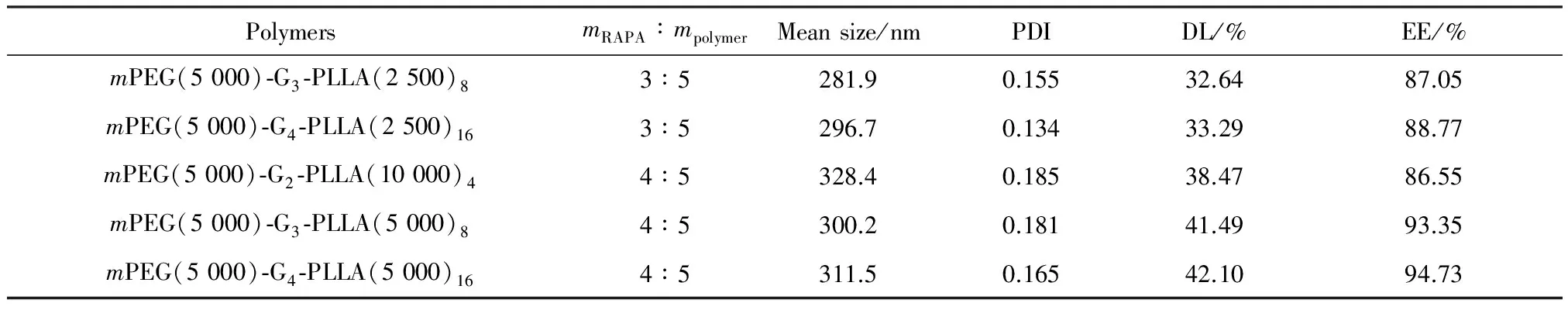
表3 不同結構線性-樹枝狀共聚物載藥微球的性能對比
從表3的數據可以看出,當共聚物的代數相同時,單鏈PLLA相對分子質量小,相應的載藥微球的載藥量、包封率和粒徑也小;mPEG(5 000)-G3-PLLA(5 000)8和mPEG(5 000)-G2-PLLA(10 000)4的PLLA分子總量是一樣的,但兩者載藥量等仍有差別,說明載藥量和包封率主要隨PLLA鏈長和代數的增加而增大。
線性-樹枝狀共聚物載藥微球與先前所制備的相同代數和PLLA鏈長的啞鈴型三嵌段兩親共聚物(聚L-丙交酯-dendron-聚乙二醇-dendron-聚L-丙交酯)的載藥微球[10]相比,兩者的載藥量和包封率基本相同,但線性-樹枝狀共聚物載藥微球的平均粒徑在300 nm左右,而后者的平均粒徑約為400 nm,平均粒徑有較明顯的減小,分布更均勻,而且聚乙二醇鏈段在溶液中的自由伸展使其更加穩定[12],在臨床應用上也更具優勢。
2.2 雷帕霉素載藥微球的體外釋放研究
以mPEG(5 000)-G4-PLLA(5 000)16共聚物制備的載藥微球為例,考察了不同載藥量的載藥微球在37 ℃、pH=7.4的PBS緩沖液中的藥物釋放速率P(t)隨時間t的變化關系,結果見圖3。
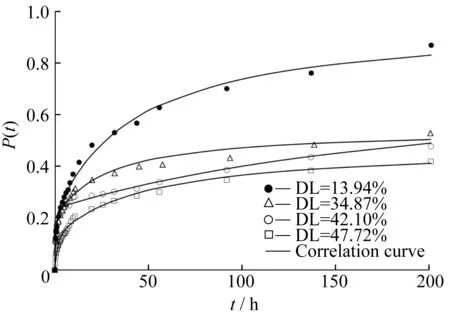
圖3 不同載藥量的載藥微球體外釋放曲線
聚合物載藥微球中的藥物釋放一般分為3階段[13]:第1階段是突釋,往往是沒有被包裹的藥物顆粒或是黏附在微球表面的藥物;第2階段是一個緩慢釋放階段,藥物穿過聚合物內核向外擴散,速度較緩慢;第3階段是快速釋放,有時也稱2次突釋,聚合物的降解或溶蝕加快藥物釋放。
在藥物初始釋放的0~3 h時間段內為突釋階段,吸附在載體表層中的藥物快速溶出,使得釋放率急劇上升,不同載藥量微球的釋放率相差不大。然后進入緩釋階段,藥物釋放量減少,速率變緩,或近似于線性變化。由于形成微球的聚合物結構相同,粒徑相當,故載藥量越低,釋放出的藥物相對量也越高,所以緩釋的速率也越快。從圖3中可以看出實驗測得的釋放率隨載藥量的增大而降低,且呈較好的遞變規律。
圖4給出了載藥量相近的線性-樹枝狀共聚物mPEG(5 000)-G3-PLLA(5 000)8和mPEG(5 000)-G4-PLLA(5 000)16兩種載藥微球釋放曲線的比較。線性-樹枝狀共聚物代數越高,聚乳酸的含量也越高,相應的粒徑也稍大些,且微球壁也越致密,使得疏水藥物RAPA與聚乳酸間的相互作用也增強[14],造成藥物在微球壁中的擴散速率變緩,累計釋放量降低。聚乳酸鏈段長度相同的情況下,代數越高,聚乳酸的含量也越多,釋放速率就越慢。
圖4給出了與mPEG(5 000)-G3-PLLA(5 000)8和mPEG(5 000)-G4-PLLA(5 000)16結構相對應的啞鈴型三嵌段兩親共聚物所制備的載RAPA微球的釋放曲線,其聚乳酸含量更高,粒徑也相應增大,故緩釋階段的釋放速率更慢。而線性-樹枝狀共聚物載藥微球在緩釋階段的緩釋速率斜率就大得多,說明其微球致密度低,因而藥物向外擴散的速率也較快。
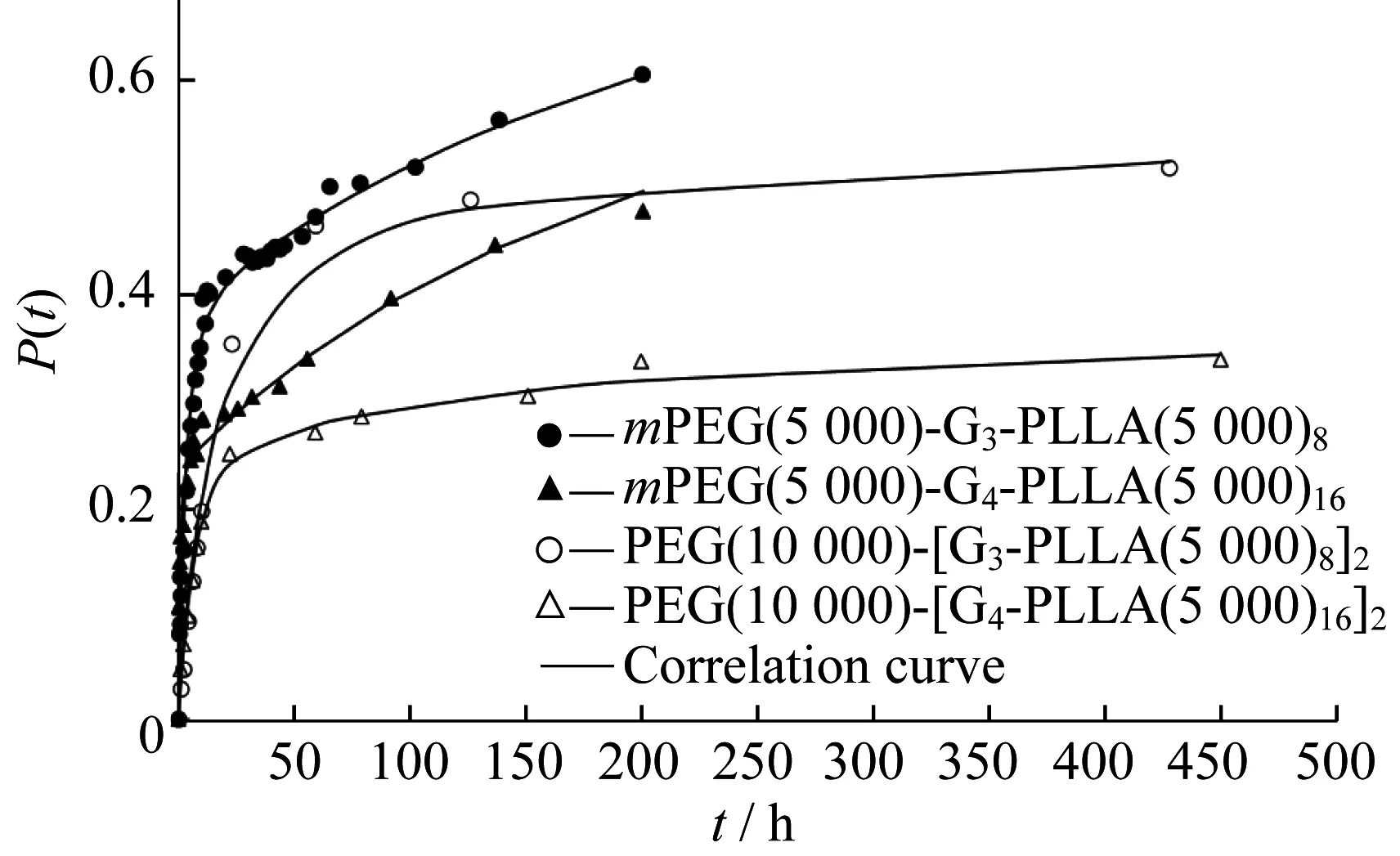
圖4 不同結構的共聚物所制備的載藥微球體外釋放曲線的比較
本課題組曾以Gompertz一級函數作為藥物釋放動力學模型來描述啞鈴型三嵌段兩親共聚物載藥釋放曲線[10],發現其能較好地描述藥物突釋階段,但隨著時間的延長,藥物仍在釋放,Gompertz一級函數模型擬合曲線不再增長,為一水平線,不能真實反映后期藥物釋放規律。對于此類微球藥物釋放情況,Gallagher等[15]提出以兩個模型的線性組合來擬合,Balcerzak等[16]也采用組合模型來擬合藥物釋放曲線,并得到了較好的效果。經考察,對于本文的研究體系,可以分別應用Gompertz一級函數和二級釋放速率方程來描述突釋階段和擴散釋放的釋放速率,將兩者線性組合的藥物釋放速率方程的形式為:
(3)
式中:P為藥物釋放率,0≤P≤1;α1、α2和β1為擬合參數;x為兩函數的組合分數;t為時間。
表4列出了采用方程(3) 關聯得到的參數、平均絕對誤差(AAE)以及平均相對誤差(ARE)。從表中可以看出,除了啞鈴型三嵌段兩親共聚物PEG(10 000)-[G3-PLLA(5 000)8]2外,擬合的效果都較好。從圖3和圖4中也可以看到,關聯曲線與實驗點也能很好地相符,且能反映出其變化趨勢。
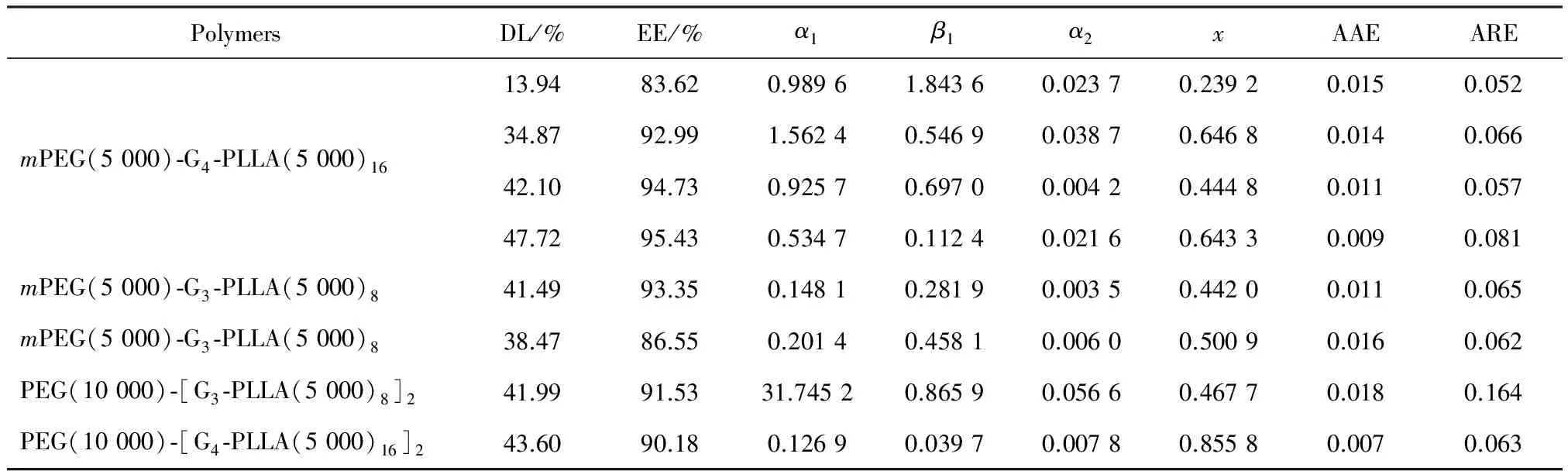
表4 方程(3)的關聯結果
3 結 論
以生物可降解的新型線性-樹枝狀兩嵌段共聚物材料作為藥物載體,采用透析法制備了RAPA載藥微球,與先前的啞鈴型三嵌段兩親共聚物載藥微球相比,保持了40%以上的高載藥量及90%以上的包封率,但平均粒徑大大減小,只有300 nm左右。其體外釋放行為與載體材料的結構有關。可以通過設計藥物載體的結構,來控制RAPA載藥微球的釋放速率,具有潛在的臨床應用價值。
[1]LI Chuan,ZHANG Jia,ZU Yujiao,etal.Biocompatible and biodegradable nanoparticles for enhancement of anti-cancer activities of phytochemicals[J].Chinese Journal of Natural Medicines,2015,13(9):641-652.
[2]KIM J,WILSON D R,ZAMBONI C G,etal.Targeted polymeric nanoparticles for cancer gene therapy[J].Journal of Drug Targeting,2015,23(7-8):627-641.
[3]商宏華,申有青.血清白蛋白納米藥物載體的制備及應用[J].功能高分子學報,2013,26(3):317-324.
[4]婁芳慧,袁金芳,高青雨.Ch-PCL-b-PVP生物可降解膠束的制備及其對布洛芬的控制釋放[J].功能高分子學報,2013,26(4):410-416.
[5]ELSABAHY M,WOOLEY K L.Design of polymeric nanoparticles for biomedical delivery applications[J].Chemical Society Reviews,2012,41(7):2545-2561.
[6]HAN Hwa Seung,LEE Jungmin,KIM Hyun Ryoung,etal.Robust PEGylated hyaluronic acid nanoparticles as the carrier of doxorubicin:Mineralization and its effect on tumor targetability in vivo[J].Journal of Controlled Release,2013,168(2):105-114.
[7]李晨晨,龔飛榮,程樹軍,等.姜黃素緩釋膠束的制備、表征及其抗細胞毒類藥物的多藥耐藥性評價[J].華東理工大學學報(自然科學版),2014,40(5):562-567.
[8]WANG Jian,CHENG Shujun,Gong Feirong,etal.Synthesis and study of biomaterials of modified poly(ethylene glyco1) with dendrimers[J].New Chemical Materials,2009,37(9):58-60.
[9]謝威,龔飛榮,江琳,等.樹枝狀聚L-丙交酯-dendron-聚乙二醇-dendron-聚L-丙交酯的自組裝[J].華東理工大學學報(自然科學版),2010,36(4):529-534.
[10]江琳,龔飛榮,范霄宇,等.雷帕霉素緩釋膠束的制備及其釋放行為[J].華東理工大學學報(自然科學版),2012,38(1):23-27.
[11]郭睿,陳明宇,范霄宇,等.生物可降解啞鈴狀高分子共聚物替莫唑胺載藥微球的制備與表征[J].華東理工大學學報(自然科學版),2014,40(5):555-561.
[12]惠志倩,郭睿,龔飛榮,等.線性-樹枝狀兩親聚合物及其膠束的制備和聚集行為[J].華東理工大學學報(自然科學版),2016,42(6):800-807.
[13]FREDENBERGA S,WAHLGRENB M,RESLOWC M,etal.The mechanisms of drug release in poly(lactic-co-glycolic acid)-based drug delivery systems:A review[J].International Journal of Pharmaceutics,2011,415(1-2) 34-52.
[14]WU Yan,WANG Tiewei,LI Mingjun,etal.Hyperbranched poly(amine-ester)-poly(ε-caprolactone) copolymer and their nanoparticles as camptothecin delivery system[J].Journal of Polymer Research,2011,18(5):1147-1158.
[15]GALLGHER K M,CORRIGAN O I.Mechanistic aspects of the release of levamisole hydrochloride from biodegradable polymers[J].Journal of Controlled Release,2000,69(2):261-72.
[16]BALCERZAK J,MUCHA M.Analysis of model drug release kinetics from complex matrices of polylactide-chitosan[J].Progress on Chemistry and Application of Chitin and Its Derivatives,2010,15:117-125.
Preparation,Characterization and Release Behavior of Drug-Loaded Microparticles by Linear-Dendritic Diblock Copolymer
YANG Li-ying1, GUO rui2, RONG Zong-ming1
(1.School of Chemisty and Molecular Engineering,East China University of Science and Technology, Shanghai 200237,China;2 BASF Shanghai Coatings Co. Ltd,Shanghai 201108,China)
The rapamycin-loaded spherical microparticle based on biodegradable linear-dendritic diblock copolymer is prepared by dialysis method.Scanning electronic microscope(SEM),dynamic light scattering(DLS) and UV spectrophotometer were used to investigate morphology,size andinvitrorelease behavior of these particles.The results showed that the drug content and encapsulation efficiency of drug-loaded microparticles prepared by diblock copolymer,as well as those prepared by dumbbell-shaped triblock copolymer,could be more than 40% and 90%,respectively.Meanwhile,the mean size of those microparticles reduced significantly and it showed an obvious sustained release as well.
linear-dendritic diblock copolymer; biodegradable; drug-loaded microparticles; druginvitrorelease; rapamycin
1006-3080(2017)01-0050-06
10.14135/j.cnki.1006-3080.2017.01.008
2016-04-27
楊李影(1989-),女,安徽阜陽人,碩士生,研究方向為界面與膠體化學。E-mail:liyingyang2014@126.com
戎宗明,E-mail:rongzm@ecust.edu.cn
O631;R318.08;R944.5
A
